
Las redes sociales nos han dado una ventana al interior de la vida de otras personas: dónde viajan, cómo visten, restaurantes de moda… es un catálogo online. Idealizamos sobre todo a aquellas personas que muestran un nivel económico mucho superior al nuestro. Personas que llevan gafas de sol personalizadas, zapatos de 650€, bolsos de marca por valores que rondan los 20.000€, zapatos, joyas, cirugías estéticas, jets privados… la lista es interminable. Lo vemos como caprichos no esenciales, pero, ¿y si le damos la vuelta? ¿Y si ahora esos precios se pagaran por aumentar la esperanza de vida de otra persona? ¿Por tratar una enfermedad con mortalidad alta? En este caso, ¿te parecería excesivo un precio de 2 millones de dólares por un frasco de vidrio? Exactamente 2.1 millones de dólares es el precio de Zolgensma, un medicamento que se emplea para el tratamiento de la atrofia muscular espinal. Afortunadamente, el Ministerio de Sanidad de España lo ha incluido en la prestación farmacéutica del sistema nacional de salud para los pacientes con esta enfermedad.
La Atrofia Muscular Espinal (AME) engloba una serie de enfermedades genéticas caracterizadas por la debilidad muscular que puede llegar a afectar al habla, respiración y movimiento como consecuencia de la descomposición de las motoneuronas.
Entre las enfermedades neuromusculares, la AME es la segunda enfermedad más común, teniendo una prevalencia de 2 afectados por cada 100.000 personas. Concretamente en España, se estima que entre 800 y 1000 personas padecen Atrofia Muscular Espinal.
Historia y herencia
En 1891 fueron realizadas las primeras observaciones de AME por Guido Werdnig y en 1990 se determinó que la causa de la enfermedad se localizaba en el cromosoma 5. Pero no fue hasta cinco años después que, gracias a Dra. Judith Melki, se localizó la lesión genética que produce la AME. Dicha lesión, presente en el 95% de los enfermos, corresponde a la pérdida o deleción de un pequeño fragmento de ADN del gen SMN1.
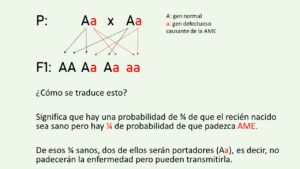
Al tratarse de un trastorno autosómico recesivo, esta pérdida de ADN está presente tanto en el cromosoma heredado de la madre como en el del padre. Es decir, ambos padres han de tener una copia del gen defectuoso para transmitírsela a la descendencia. En el caso de ser los dos padres portadores, la probabilidad de tener un hijo enfermo sería del 25%.
Sabiendo ya cómo se hereda el gen defectuoso, vamos a ver con detalle las características de este gen pero sobre todo, de la proteína que codifica.
La proteína SMN
La proteína SMN (Survival Motor Neuron), como su nombre indica, es necesaria para la supervivencia de las neuronas motoras. Esta proteína se expresa en todos los tejidos y se localiza tanto en el citoplasma como en el núcleo celular. Así pues, ¿qué son las motoneuronas? Las motoneuronas son neuronas especializadas en la transmisión del impulso nervioso a los músculos. Es decir, se encargan de transformar la voluntad de, por ejemplo, mover el brazo, en movimiento.
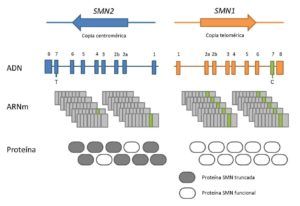
La proteína SMN está codificada por dos genes: SMN1, el cual produce la proteína completa y SMN2, que produce una pequeña cantidad de la proteína completa pero principalmente produce una forma truncada. Cuando se producen mutaciones en el gen SMN1, la cantidad de proteína codificada prácticamente desaparece, ya que los niveles de proteína que puede proporcionar SMN2 no son suficientes. Sí se ha observado que, por regla general, cuantas más copias del gen SMN2 tiene el paciente, la enfermedad es menos grave.
Con todo esto se puede concluir que la atrofia muscular espinosa ocurre como consecuencia de la carencia de la proteína SMN completa, y que su grado de gravedad está influenciado principalmente por la complementación del gen SMN2.
Pero, ¿qué ocurre exactamente cuando los niveles de SMN son bajos?
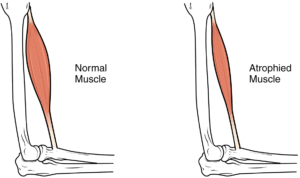
No se sabe con certeza por qué las motoneuronas son tan sensibles a los niveles bajos de SMN. Existen diferentes hipótesis al respecto. La más aceptada sugiere que la deficiencia de SMN provoca que no se transcriban correctamente una serie de proteínas necesarias para el adecuado funcionamiento de las motoneuronas. Como consecuencia, los músculos se quedan sin la estimulación necesaria para ser funcionales, por lo que acaban perdiendo fuerza y, finalmente, atrofiándose.
Tipos de Atrofia Muscular Espinal
Como hemos comentado antes, hay 4 tipos diferentes de atrofia. Todos tienen en común defectos en el gen SMN1 y su gravedad está determinada por la cantidad de copias del gen SMN2, siendo el tipo I el más grave con el porcentaje de mortalidad más elevado y por lo tanto, el que menos copias del gen SMN2 presenta.
Cada tipo de AME manifiesta unas características diferentes.
|
Tipo I |
Tipo II | Tipo III |
Tipo IV |
|
| Aparición de los síntomas | 6 meses | 6-18 meses | Después de los 18 meses, entre 2-17 años |
A partir de los 21 años |
|
Características |
Los niños afectados no pueden sentarse ni pararse | Pueden sentarse sin apoyo pero no pueden caminar sin ayuda | Pueden caminar solos aunque pueden perder esta capacidad progresivamente
Tienen problemas para correr o subir escaleras |
Vida normal hasta la aparición de los síntomas |
|
Síntomas |
Disminución del tono muscular, dificultad para tragar y respirar | Problemas respiratorios | Pueden presentar escoliosis, contracturas e infecciones respiratorias |
Debilidad moderada en la musculatura de las piernas, temblores y problemas respiratorios leves |
|
Frecuencia |
Más común, entre el 50- 60% de los casos | 30% de los casos | 10-20% de los casos | Poco común entre el 1-5% de los casos |
|
Gravedad |
Alta | Moderada/alta | Leve |
Leve |
|
Esperanza de vida |
Sin tratamiento, morirán antes de los 2 años | Reducida pero con tratamiento, pueden llegar a adolescentes | Con tratamiento tienen una expectativa de vida normal |
Expectativa de vida normal |
|
Copias del gen SMN2 |
1-2 | 3-5 | 3-5 |
|
¿Cómo se diagnostica la atrofia muscular espinal?
Como consecuencia de que cada enfermo presenta signos y síntomas a diferentes edades y con diferente gravedad, es posible que el diagnóstico final se retrase. Hemos visto los síntomas asociados a cada tipo de AME anteriormente, pero para confirmar la presencia de la enfermedad, el procedimiento habitual es la realización de un análisis genético. Se toma una muestra de sangre y se analiza si contiene, o no, una mutación específica en el gen SMN1. Mediante este mismo análisis se pueden determinar también las copias de gen SMN2 existentes.
Este análisis puede hacerse también antes de que el bebé nazca si se han confirmado antecedentes familiares. Se realizará entonces una amniocentesis, que consiste en el análisis de una muestra de líquido amniótico. Esta prueba si el gen SMN1 está o no mutado.
En la mayoría de los casos, entre un 90-95%, el problema está causado por las mutaciones en este gen. En los casos en los que no se encuentran cambios genéticos, hay otros test que pueden llevarse a cabo para determinar la presencia de la enfermedad como son exámenes físicos, electromiografías, estudios de conducción nerviosa o biopsias musculares.
¿Cuáles son los tratamientos para la atrofia muscular espinal?
Existen dos formas de encarar el tratamiento: una dirigida a resolver la falta de proteína característica de la enfermedad y otra encaminada a convivir con ella.
En el primer caso, se sigue investigando pero hay algunos tratamientos disponibles que pueden ayudar a controlar los síntomas y prevenir complicaciones. Por ejemplo, encontramos un medicamento cuya ingesta aumenta los niveles de proteína SMN. Concretamente el Risdiplam incrementa los niveles de la proteína gracias a regular la expresión de la proteína SMN a partir del gen SMN2.
El último tratamiento en salir al mercado es Zolgensma, un medicamento sólo indicado para menores de 2 años que presentan la tipología I de AME. Se administra una única dosis de forma intravenosa. El objetivo final de este tratamiento es reparar los genes dañados para que sean capaces de producir la proteína SMN en cantidades normales. ¿Cómo lo logran? Zolgensma está basada en adenovirus genéticamente modificados que actúan como transportadores de una copia corregida del gen SMN1, buscando que este gen modificado se exprese en las motoneuronas.
A pesar de los prometedores resultados que está teniendo, este medicamento ha levantado mucha polémica como consecuencia de su elevado precio: 2.1 millón de dólares la dosis.
Como habréis podido comprobar, los tratamientos disponibles (Risdiplam y Zolgensma) dirigidos a recuperar la proteína SMN funcional no son completamente efectivos. Y todos tienen en común que no curan la AME de forma definitiva, ya que es una enfermedad que no tiene cura, a día de hoy. Por ello se siguen utilizando tratamientos paliativos dirigidos a tratar los síntomas. El nivel de cuidados lo determinan los padres (en casos de niños afectados) y médicos en conjunto. Dichos cuidados se abordan en 3 flancos diferentes e incluyen:
- Cuidados respiratorios: en los casos en los que la debilidad muscular afecta a la capacidad respiratoria son necesarios respiradores mecánicos que envían un flujo de aire a pulmones. También existen dispositivos de tos asistida, para ayudar con la expulsión de secreciones y mucosidades. En los casos más graves, son necesarias las intubaciones o traqueotomías
- Nutrición: cuando la musculatura implicada en la masticación y deglución de alimentos está afectada, la forma de alimentación es mediante sondas nasogástricas, que entran a través de la nariz y transportan la comida hasta el estómago. Es importante tener en cuenta que estos niños pueden llegar a desarrollar desnutrición o neumonías (si aspiran alimentos o líquidos mientras comen). Sorprendentemente, también pueden desarrollar obesidad si ingieren demasiadas calorías para su nivel de actividad, por lo que son necesarias visitas periódicas a nutricionistas para evitar cualquiera de estas situaciones.
- Fisioterapia: el objetivo es llevar vidas lo más normales posibles a pesar de las limitaciones físicas. Se han observado muchos casos de mejora gracias a la asistencia periódica a fisioterapia. Recordemos que el problema principal de esta enfermedad es la atrofia de los músculos como consecuencia de la descomposición de las motoneuronas. La fisioterapia les ayuda a favorecer la circulación sanguínea en las extremidades y a aliviar el dolor consecuencia de la debilidad e inmovilidad muscular. También se hacen ejercicios para tonificar o reforzar los músculos que participan en la respiración, además de practicar ejercicios que les ayudan en actividades cotidianas como andar, vestirse, peinarse y cepillarse los dientes con toda la independencia posible. Los fisioterapeutas, a través de la realización de todos estos ejercicios, ayudan a mejorar la calidad de vida de estos niños. También se disponen de instrumentos que facilitan algunas tareas como férulas para las piernas, bipedestadores o bastones.
Fuentes:
Fundación atrofia muscular espinal https://www.fundame.net/
Instituto nacional de trastornos neurológicos y accidentes cerebrovasculares: https://espanol.ninds.nih.gov/es
Together in SMA https://care.togetherinsma.es/es_ES/home.html
Roche pacientes https://rochepacientes.es/
Medline https://medlineplus.gov/spanish/
Kids Health https://kidshealth.org/
Fisio Online https://www.fisioterapia-online.com/
Tesis doctoral. Laura Torres Benito. Función de la proteína de supervivencia de motoneuronas en la maduración funcional y organización sináptica en un modelo murino de atrofia muscular espinal. 2012. https://idus.us.es/bitstream/handle/11441/40896/S_TD_PROV178.pdf?sequence=4
Si te ha gustado este blog y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria, así como nuestro canal audiovisual, Genotipia TV.



