En el siglo XVII, concretamente en el año 1686, Thomas Sydenham, médico inglés al que se le atribuyó el sobrenombre de “ Hipócritas Británico”, describió una extraña dolencia a la que denominó “chorea sancti viti”. Desde ese momento, hasta nuestros días, la expresión “baile de San Vito” se ha utilizado para hacer referencia a un nutrido conjunto de trastornos neurológicos, caracterizados por la presencia de movimientos involuntarios incontrolados. El más representativo de todos ellos es la enfermedad de Huntington.
La enfermedad de Huntington se desarrolla en base a una alteración genética, concretamente a una expansión de trinucleótidos. Por ello, en nuestro post de hoy, elaboraremos un recorrido a través de las principales enfermedades causadas por expansiones de trinucleótidos.
El incremento en el número de repeticiones de ciertas secuencias de trinucleótidos del ADN, es la base molecular de un número creciente de enfermedades multisistémicas, especialmente neurológicas y neurodegenerativas. Existen cuatro tipos de tripletes cuya expansión resulta especialmente patogénica en seres humanos: CGG/GCC, CAG/GTC, CTG/CAG y GAA/CTT. Las expansiones nucleotídicas pueden estar presentes tanto en secuencias codificantes como en secuencias no codificantes.
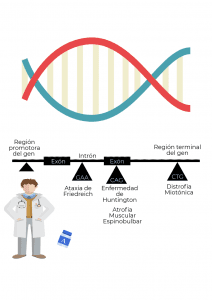
Las expansiones de trinucleótidos pueden causar diferentes efectos: desde la inactivación de un gen hasta alteraciones en el transporte del ARN mensajero y/o en la síntesis proteica. Además, las patologías que tienen su origen en la expansión y/o repetición de trinucleótidos presentan características diferenciales, en cuanto a su cuadro clínico y pronóstico, por lo que. A continuación, explicaremos de manera detallada las características de las más relevantes.
Enfermedad de Huntington
La enfermedad de Huntington, patología con la que iniciábamos el recorrido a través de las principales enfermedades causadas por expansión de trinucleótidos, es una alteración de carácter autosómico dominante.
El principal gen relacionado con la aparición de esta enfermedad es el gen HTT, situado en el brazo corto del cromosoma 4. Este gen, aislado en el año 1993, presenta una región polimórfica de repeticiones CAG en el exon 1. En personas sanas, el número de repeticiones es inferior a 26. Mientras que en personas afectadas por la enfermedad los valores se sitúan en el intérvalo de las 36 – 121 repeticiones.
En el caso de la enfermedad de Huntington, la amplificación de trinucleótidos se produce preferentemente por vía paterna y está asociada al fenómeno de anticipación genética.
El transcrito del gen HTT relacionado con la enfermedad se localiza en el cerebro y otros tejidos y produce una proteína denominada huntingtina. En los pacientes afectos con esta enfermedad, el exceso en el número de repeticiones del triplete CAG resulta en una versión de la proteína huntingtina demasiado grande e inestable, que se escinde generando fragmentos que resultan tóxicos para las neuronas. Esta toxicidad deriva, a su vez en la generación de la sintomatología típica de la enfermedad.
Por lo que respecta a la sintomatología, en el curso de la enfermedad de Huntington convergen alteraciones cognitivas, psiquiátricas y motoras. La sintomatología concreta varía de una persona a otra en función de la amplitud, severidad, edad de comienzo y la velocidad de progresión. Sin embargo existen una serie de síntomas característicos comunes como son los movimientos involuntarios incontrolados y alteraciones musculares.
Distrofia miotónica tipo 1
La distrofia miotónica constituye la miopatía más frecuente en adultos. Se trata de una patología de carácter dominante. El principal gen implicado en su patogénesis es el gen DMKT, cuyo transcrito se ha localizado en el músculo esquelético y cardíaco. Este transcrito codifica para una proteína que se localiza en las uniones neuromusculares.
El proceso etiopatogénico de la distrofia miotónica de tipo 1 se origina a partir de la expansión del trinucleótido CTG en la región 3’UTR del gen DMKT. Tras la transcripción de este gen, se forman en el ARN horquillas que resultan tóxicas. Estas horquillas son capaces de secuestrar proteínas relevantes en los procesos de splicing alternativo, como MBNL. La ausencia de MBNL, provoca alteraciones en el metabolismo del ARN que desencadenan los síntomas de la enfermedad.
En condiciones normales, el número de repeticiones de este triplete se sitúa comprendido en el intérvalo entre 5-34 y las premutaciones (entre 35 y 49 repeticiones) no causan patología. Sin embargo, son inestables a la hora de transmitirse y son capaces de expandirse en el rango patológico en la siguiente generación. Un número de repeticiones superior al valor 50 resulta patológico.
Esta patología multisistémica presenta un cuadro clínico muy variable. La afectación es principalmente neurológica, con debilidad muscular, atrofia y miotonía como principales síntomas de la enfermedad. Sin embargo, también se ven afectados otros sistemas, causando síntomas tan dispares como alteraciones en la conducción cardíaca, cataratas, hipersomnia, atrofia testicular y calvicie prematura en varones.
Ataxia de Friedreich
La Ataxia de Friedreich es una patología neurodegenerativa con patrón de herencia autosómico recesiva. El origen de la misma es una anomalía en el gen FXN, concretamente una repetición excesiva del trinucleótido GAA. En condiciones normales, este trinucleótido se presenta en aproximadamente 8-30 repeticiones, mientras que, en condiciones patológicas puede alcanzar las 1000 repeticiones.
El gen FXN, codifica para la proteína frataxina. La expansión aberrante de este trinucleótido provoca una perturbación a nivel transcripcional, que deriva en una reducción de la cantidad de frataxina producida. Esta reducción ocasiona que algunas células del cuerpo, concretamente, las neuronas, células de la médula espinal y musculares, no sean capaces de soportar la elevada tensión oxidativa generada por las mitocondria dando lugar al cuadro clínico característico de la enfermedad.
La sintomatología típica se relaciona con el desgaste en la estructura de zonas cerebrales y espinales, que controlan la coordinación, el movimiento muscular y otras funciones. Los principales síntomas son la dificultad para caminar, disminución de la sensibilidad, debilidad muscular, así como, anomalías rítmicas del corazón.
Atrofia muscular espinobulbar
La atrofia muscular espinobulbar es una patología neuromuscular que presenta un patrón de herencia recesiva ligada al cromosoma X. Está causada por una expansión inestable de la repetición del triplete CAG en el exón 1 del gen receptor de andrógenos (AR) en el cromosoma Xq11-12.
En condiciones normales, el trinucleótido se encuentra presente en un número de copias situado en torno a 10 – 36 repeticiones. Por otro lado, en personas afectas el rango de de variabilidad de las repeticiones se sitúa en torno a las 40 – 52 repeticiones. En el caso de las personas afectadas, este exceso de repeticiones provoca alteraciones estructurales en la proteína codificada por este gen, lo que, a su vez, se traduce en alteraciones funcionales en las neuronas motoras del cerebro y de la médula espinal. Las neuronas afectadas mueren actuando como detonante de la aparición de los síntomas de la enfermedad.
La atrofia muscular espinobulbar se caracteriza por una debilidad muscular progresiva, atrofia y fascilulaciones en la musculatura proximal y bulbar.
Anticipación genética
La anticipación genética es un fenómeno característico de algunas enfermedades entre las que destacan las enfermedades causadas por expansión de trinucleótidos que nos ocupan. Este fenómeno consiste en la existencia de una relación directamente proporcional entre el número de repeticiones adicionales del trinucleótido y el momento de aparición de la enfermedad y/o severidad de su sintomatología. Lo cual significa que, a medida que transcurren las generaciones, lo hace también el número de repeticiones, como consecuencia de lo cual, la enfermedad aparece a edades más tempranas y de forma más severa.
Para concluir, cabe señalar que, dada la incidencia y repercusiones de este tipo de patologías causadas por expansiones de trinucleótidos, se hace fundamental el estudio exhaustivo de los procesos de mutación dinámica que subyacen a este tipo de patologías.
Bibliografía:
-
Bates GP. History of genetic disease: the molecular genetics of Huntington disease – a history. Nat Rev Genet. 2005 Oct;6(10):766-73.
-
Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009 Mar;256 Suppl 1:9-17. doi: 10.1007/s00415-009-1003-2. Review.
-
Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. 2005 Jan;15(1):5-16. Epub 2004 Nov 26. Review.
- Banno, H., Katsuno, M, et al. Phase 2 Trial of Leuprorelin in
Patients with Spinal and Bulbar Muscular Atrophy. Ann Neurol
2009;65:140-150.