En el ámbito de los agentes infecciosos, existe una amplia diversidad, que incluye bacterias patógenas, hongos contagiosos y persistentes virus. Sin embargo, entre todos ellos, los priones ocupan un lugar destacado por su capacidad destructiva y peculiaridad biológica. Estos silenciosos pero letales agentes proteicos son los responsables de las encefalopatías espongiformes transmisibles en mamíferos, patologías que destacan tanto por su rareza como por su gravedad.
Antes de abordar el impacto de los priones en la salud humana y los aspectos genéticos asociados, es crucial comprender qué son exactamente estos agentes infecciosos.
¿Qué son los priones?
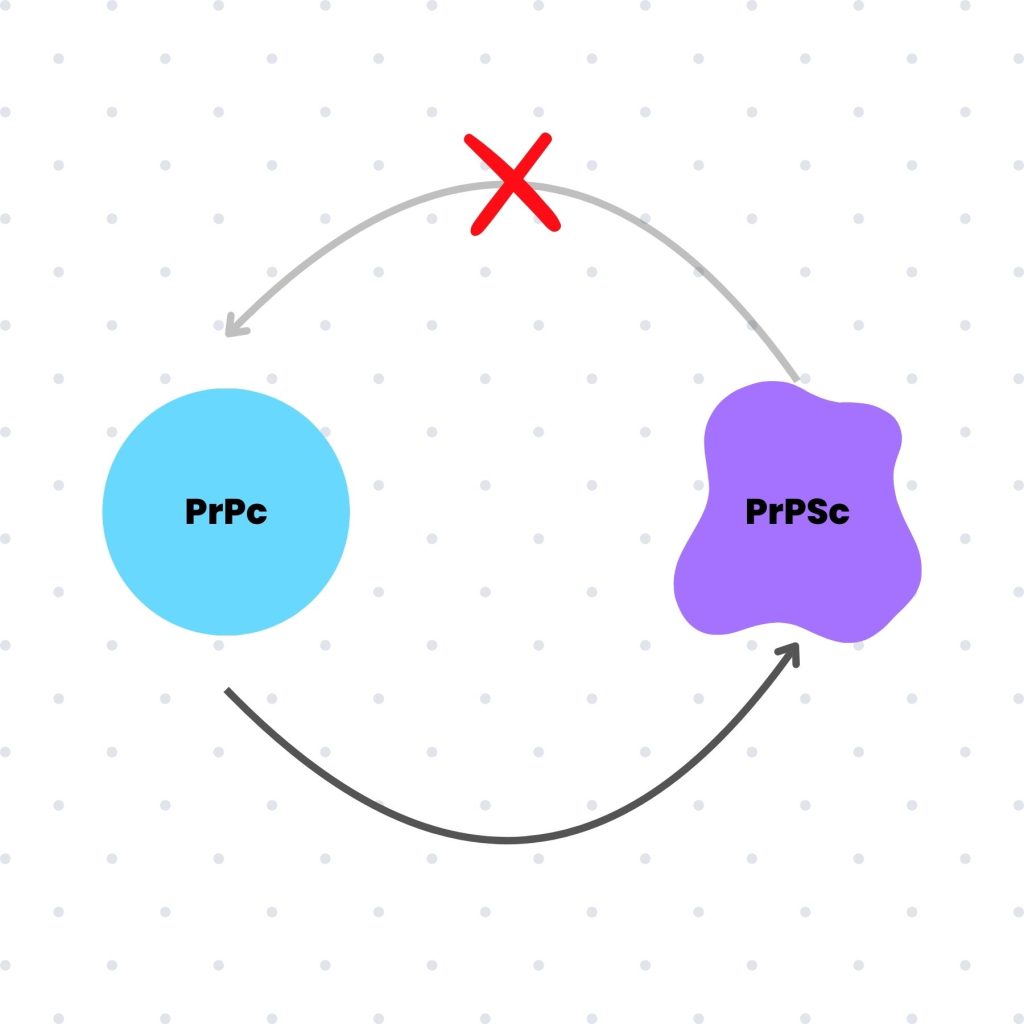
Un prión puede definirse como una proteína defectuosa, aunque este término no logra captar completamente su singularidad. Los priones no solamente son proteínas defectuosas, sino que también poseen la inquietante capacidad de inducir cambios en proteínas funcionales del organismo afectado, transformándolas en nuevas formas patógenas.
El origen de estas proteínas se encuentra en el gen PRNP, ubicado en el cromosoma 20 humano. Este gen, en su forma normal, codifica una proteína funcional conocida como PrPc, que desempeña funciones importantes en el tejido nervioso. Sin embargo, cuando esta proteína adopta la isoforma anómala PrPSc, comienza el proceso patológico.
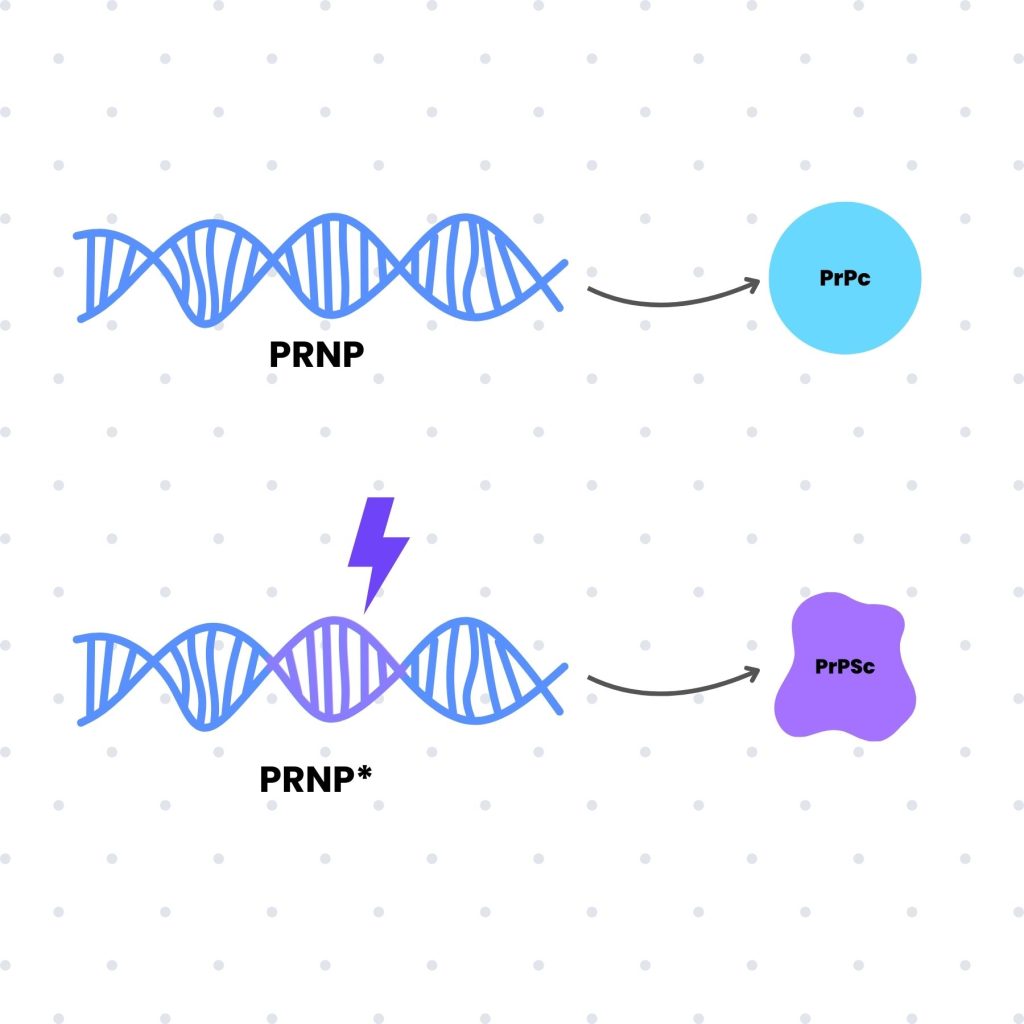
La isoforma PrPc es soluble y presenta una estructura globular que favorece su integración en el metabolismo celular. En contraste, la isoforma PrPSc adopta una conformación plana e insoluble, lo que lleva a su acumulación en forma de agregados proteicos dentro de las células nerviosas. Estos agregados interfieren en la función neuronal, desencadenando un proceso neurodegenerativo que resulta en graves alteraciones funcionales.
Lo que hace particularmente peligrosos a los priones es su resistencia a los sistemas de detección y eliminación de proteínas defectuosas en el organismo. Incluso las proteasas, encargadas de degradar proteínas anómalas, son ineficaces frente a ellos.
Enfermedades priónicas
Las enfermedades causadas por priones se agrupan bajo el término encefalopatías espongiformes transmisibles (EET). Estas patologías afectan principalmente al sistema nervioso central, produciendo una característica apariencia porosa en los tejidos afectados, debido a la neurodegeneración.
Entre los síntomas comunes se encuentran el deterioro gradual de las capacidades motoras y cognitivas, aunque la presentación clínica puede variar según la enfermedad específica.
Mecanismos de Transmisión de las enfermedades causadas por priones
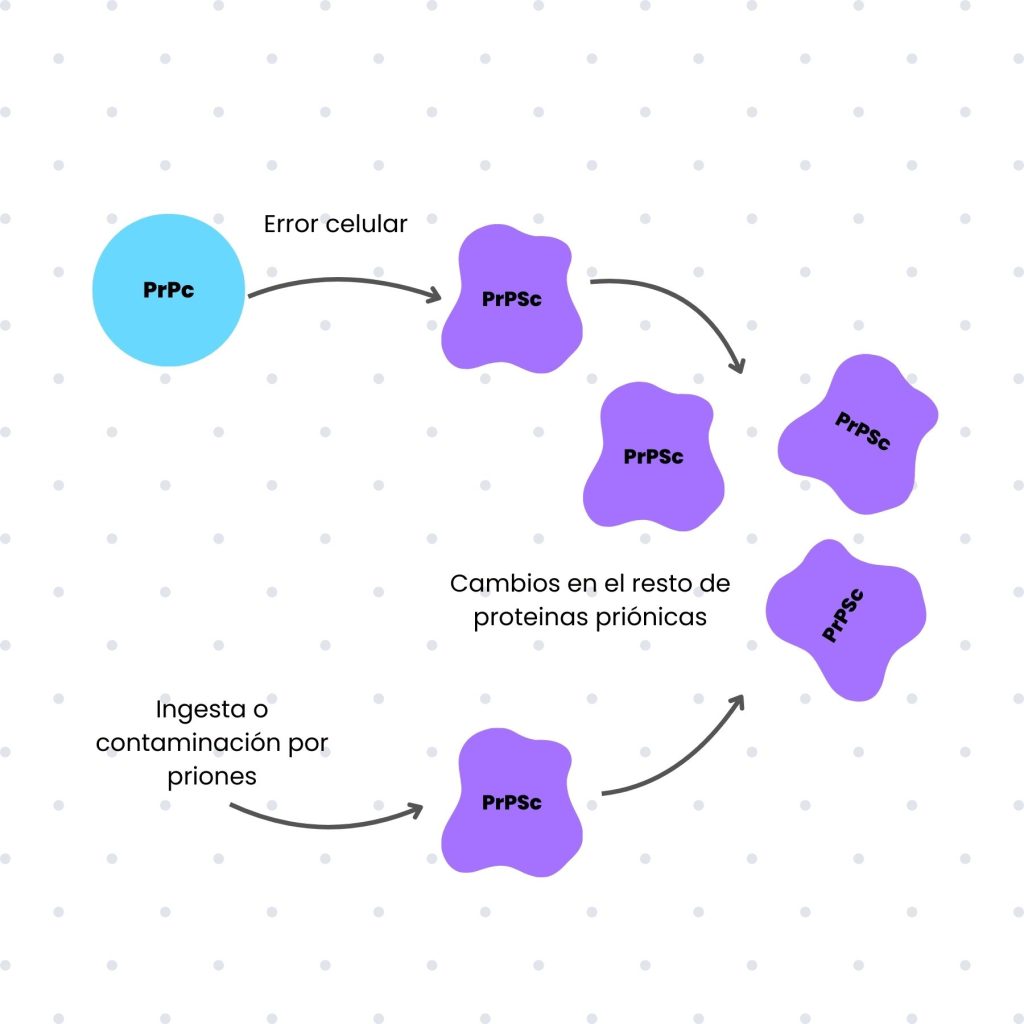
Las enfermedades priónicas representan una minoría entre las patologías infecciosas, ya que los priones carecen de mecanismos de infección activa. Por lo general, estas enfermedades son espontáneas, resultado de errores celulares que generan la primera isoforma PrPSc, la cual induce cambios en las proteínas normales.
Sin embargo, existen situaciones en las que estas patologías pueden ser transmisibles, como el caso del kuru, enfermedad asociada al canibalismo ritual practicado en Papua Nueva Guinea, o la encefalopatía espongiforme bovina (enfermedad de las «vacas locas»), vinculada al consumo de carne contaminada. También es posible la transmisión a través de procedimientos médicos realizados con instrumentos contaminados.
Enfermedades priónicas hereditarias
Aunque la mayoría de las enfermedades priónicas tienen un origen espontáneo, algunas son de carácter hereditario. Estas son la enfermedad de Creutzfeldt-Jackob familiar, el Síndrome de Gerstmann-Straüssler-Scheinker y el Insomnio familiar fatal. En estos casos, al conocerse la base genética, se puede diagnosticar a los pacientes, asesorarlos de cara a tener descendencia o informar a los familiares de sus probabilidades de desarrollar la enfermedad.
Enfermedad de Creutzfeldt-Jakob Familiar
La enfermedad de Creutzfeldt-Jackob de origen familiar es una prionopatía descrita en 4 familias del sudeste de Inglaterra. Generalmente, los individuos de estas familias comenzaban, normalmente en edad adulta, a sufrir episodios de demencia y deterioro de las capacidades cognitivas, seguidos de una constante disminución en la coordinación motora (ataxia). Además, los miembros enfermos de estas familias solían morir poco después de presentar los primeros síntomas.
La incapacidad de realizar un estudio más afinado hizo que los miembros de estas familias fuesen diagnosticados como enfermos de Alzhéimer, Huntington, Párkinson, etc, pero actualmente se conocen sus bases genéticas. Se ha comprobado que el inicio de la enfermedad ocurrió por una variante alélica en el gen PRNP, el alelo M129.
La herencia de la enfermedad de Creutzfeldt-Jackob es dominante autosómica, es decir, heredar un solo alelo mutado de PRNP es suficiente para desarrollar la enfermedad. No obstante, en individuos heterocigóticos, los síntomas aparecen más tarde y el progreso es mucho más lento. que en aquellos que han heredado dos copias mutadas.
Síndrome de Gerstmann-Sträussler-Scheinker
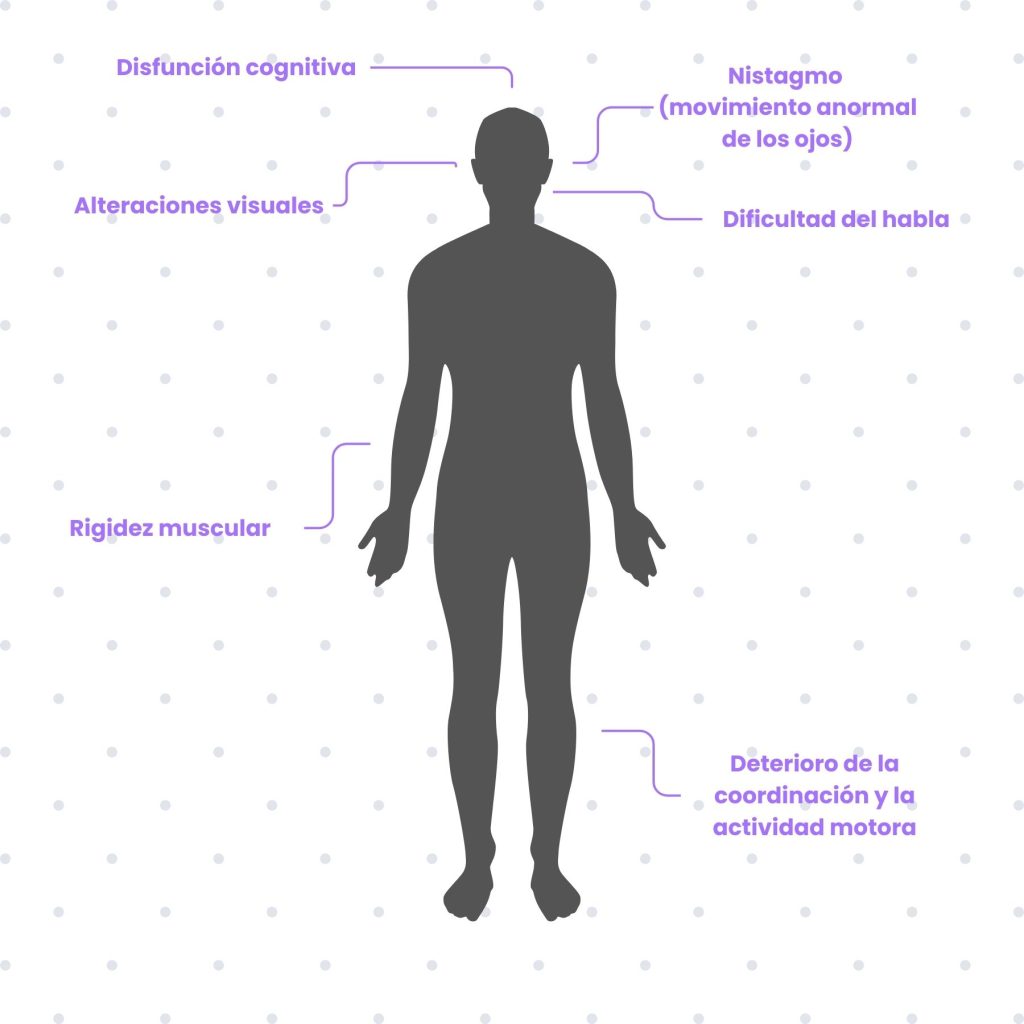
Este síndrome está causado por una variante alélica en el gen PRNP que hace que la proteína resultante tenga una estructura anormal. Su característica principal es una neurodegeneración que comienza aproximadamente sobre los 40 años y que disminuye progresivamente las capacidades motoras del enfermo. Además, los pacientes con síndrome de Gerstmann-Straüssler-Scheinker presentan disfunción cognitiva y deterioro de la coordinación y actividad motora. Se observa también dificultad del habla, alteraciones visuales, nistagmo (movimiento anormal de los ojos) y rigidez muscular.
Aunque a priori podríamos pensar que el síndrome de Gerstmann-Straüssler-Scheinker es la misma patología que la enfermedad de Creutzfeldt-Jackob, recientes análisis inmunohistoquímicos han logrado diferenciar estas dos enfermedades. Además, al contrario de lo que sucede en la enfermedad de Creutzfeldt-Jackob, los enfermos de Gerstmann-Straüssler-Scheinker comienzan a presentar los primeros síntomas a edades más tempranas (40 años en contraposición a 60). Otro punto diferencial es que los enfermos de Gerstmann-Straüssler-Scheinker presentan espasmos musculares en menor frecuencia que los enfermos de Creutzfeldt-Jackob.
Insomnio Familiar Fatal
El insomnio familiar fatal es una prionopatía hereditaria que produce insomnio severo en los enfermos, además de demencia, pánico, pérdida de peso, falta de apetito o hipotermia entre otros síntomas. Los primeros síntomas de esta enfermedad comienzan generalmente entre los 32 y 62 años.
Aunque esta enfermedad no se concentra en una única región y se han descrito casos familiares por todo el mundo (Austria, Japón, Italia, Alemania, Estados Unidos, Francia, …), en las últimas décadas se han detectado más de 40 casos en España, concretamente en la comunidad autónoma del País Vasco.
Fuentes:
https://rarediseases.info.nih.gov/espanol/13176/insomnio-familiar-fatal



