La genética puede tener un papel crucial en el desarrollo de ciertas enfermedades que afectan al sistema nervioso central. Es el caso del Huntington, una enfermedad neurodegenerativa hereditaria que, al igual que el Párkinson, provoca el deterioro de ciertas células nerviosas. En esta ocasión, os hablaremos acerca de esta enfermedad, de sus bases genéticas y de su modo de herencia. ¿Te interesa? ¡Continúa leyendo!
¿Qué es la enfermedad de Huntington?
La enfermedad de Huntington es una enfermedad genética y hereditaria que afecta a entre 5-10 personas por cada 100.000 habitantes y que se caracteriza por alteraciones motoras, cognitivas y psiquiátricas que se dan de forma progresiva. A medida que progresa la enfermedad, se observa una disminución en las funciones intelectuales. Además, se observan comportamientos de irritabilidad, impaciencia, comportamientos antisociales y episodios de psicosis, paranoia y alucinaciones.
En cuanto a alteraciones motoras, en fases avanzadas de la enfermedad se observan muecas y movimientos espasmódicos en brazos y piernas. A estas alteraciones motoras se les denomina corea de Huntington. Antiguamente se denominaba “Baile de San Vito” a la corea de Huntington (además de a la corea de Sydenham), ya que a los individuos que presentaban estos síntomas se les enviaba a peregrinar a la capilla de San Vito en Ulm (Alemania) para que el santo aliviara su padecer.

Genética de la enfermedad de Huntington
El gen afectado en los pacientes con Huntington es el HTT, que presenta una región en la que se repite, en mayor o menor medida, un triplete de bases nitrogenadas (Citosina-Adenina-Guanina). En personas sanas, el nº de repeticiones de este triplete se encuentra entre 1 y 40, mientras que las personas con Huntington suelen presentar un número de repeticiones mayor a 40.
Las proteínas sintetizadas a partir de formas del gen HTT con un alto número de repeticiones suelen precipitar formando aglomeraciones llamadas cuerpos de inclusión, que propician la degeneración neuronal en el cerebro.
Sin embargo, no todas las personas con un nº de repeticiones del triplete CAG mayor a 40 en el gen HTT desarrollarán Huntington en algún momento de su vida. De hecho, se estima que un 5% de las personas con una copia del gen mutado no presentarán la enfermedad. Esto es así porque la enfermedad de Huntington presenta penetrancia incompleta.
Herencia y anticipación genética
La herencia de la enfermedad de Huntington está ligada al cromosoma 4 (autosómica) y es de tipo dominante. Sin embargo, se observan patrones en las genealogías que no son típicos de la herencia dominante debido a un fenómeno conocido como anticipación genética.
Anticipación genética
La anticipación genética describe la tendencia de algunas enfermedades de ser más graves e incluso aparecer de forma más temprana con las generaciones; la enfermedad de Huntington es una de estas enfermedades.
Este fenómeno es debido al número de repeticiones. El gen “normal” que no produce la enfermedad de Huntington tiene menos de 40 repeticiones de estas tres bases. Se observan dos variantes dentro de esta enfermedad: la variante juvenil (+60 repeticiones) y la variante del adulto (40-60 repeticiones).
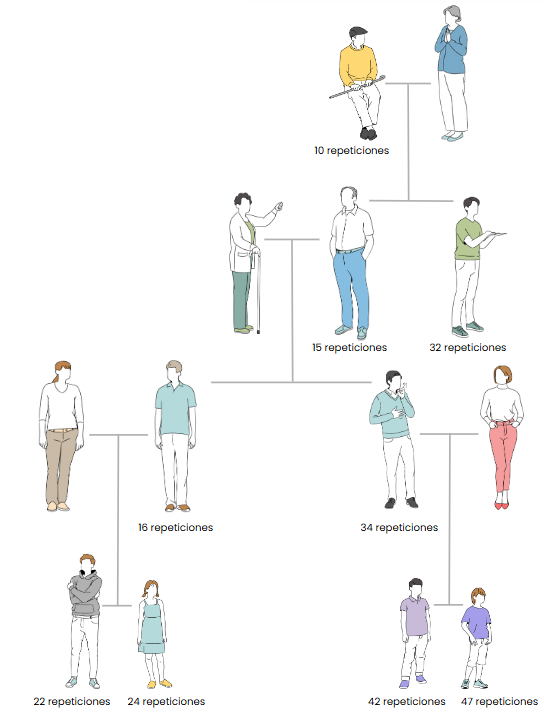
En la variante juvenil se observa una aparición muy temprana de la enfermedad (Individuos menores a 20 años), mientras que en la del adulto se observa sobre los 40-50 años.
Tratamiento en personas con Huntington
Aunque actualmente no existe un tratamiento que cure esta enfermedad o impida su progresión, existen algunos fármacos que ayudan a contrarrestar sus síntomas. Usualmente se recetan neurolépticos y bloqueantes de dopamina a aquellos pacientes que presenten trastornos motores graves. En caso de trastornos psicológicos se utilizan sedantes, antidepresivos y antipsicóticos. Estos tratamientos no mejoran el estado de la enfermedad, que continúa avanzando progresivamente, aunque sí mejoran el estilo de vida del paciente momentáneamente.
Recientemente, algunos estudios están evaluando nuevos tratamientos para revertir el progreso de la enfermedad de Huntington. Dado que esta patología está causada por la repetición de bases nitrogenadas en un único gen, es correcto pensar en la viabilidad de la terapia génica como tratamiento. En ratones se ha conseguido bloquear la expresión del gen que codifica para la huntingtina mutada utilizando fragmentos de RNA complementarios a los que se transcriben del gen.Otro tipo de aproximación es la búsqueda de moléculas capaces de traspasar la barrera hemato-encefálica y revertir los efectos de la enfermedad. Un ejemplo es un trabajo recientemente publicado en EMBO Molecular Medicine, donde un equipo de investigadores identificó dos pequeñas moléculas que consiguen reducir los niveles de huntingtina defectuosa.

