Como todos sabemos, para el correcto desarrollo del zigoto se necesita una copia del material genético, una originaria de la madre y otra del padre. Sin embargo, pocos saben que el origen materno o paterno de dicha copia puede hacer que ciertos genes se expresen de un modo específico (se activan o no dependiendo de si viene del padre o de la madre). Este fenómeno se conoce como impronta genómica.
Los síndromes de Prader-Willi y Angelman son los ejemplos más conocidos y estudiados en relación con las patologías producidas por la impronta genómica. En los dos casos se produce una microdeleción en el cromosoma 15 en una región crítica donde se encuentran genes con impronta. Ambos síndromes están afectados la misma microdeleción pero se diferencian en su origen. Si el origen del cromosoma es paterno se producirá el síndrome de Prader-Willi y si el origen del cromosoma es materno dará lugar al síndrome de Angelman.
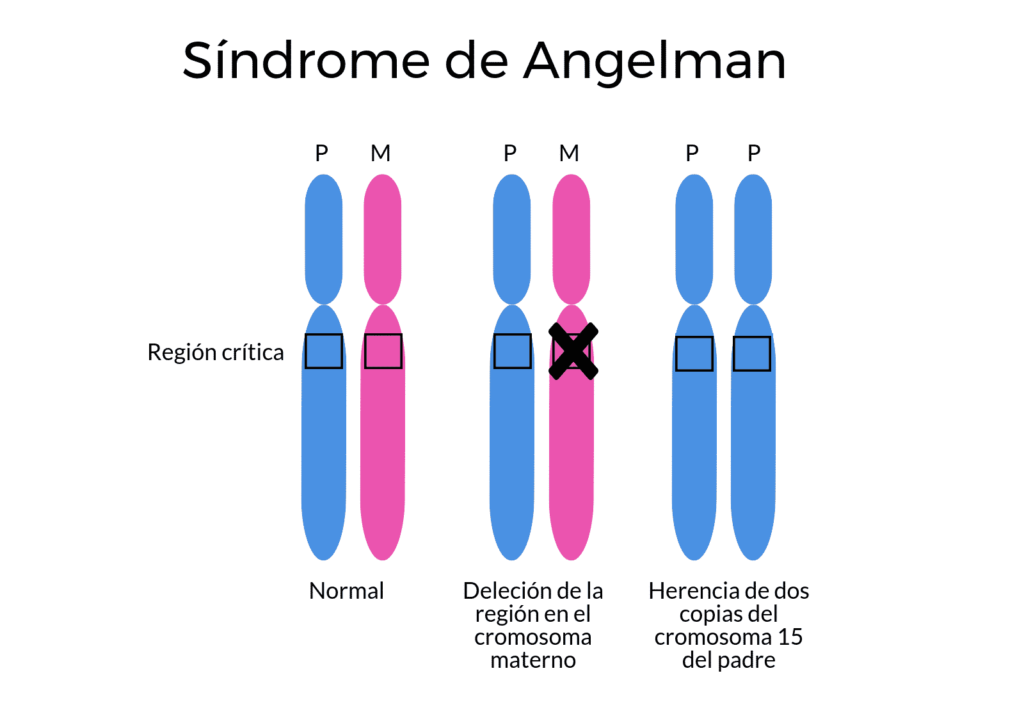
Ausencia de una copia de origen materno: síndrome de Angelman
El síndrome de Angelman es un trastorno genético que afecta principalmente al sistema nervioso. Está causado por la ausencia de una copia materna funcional del gen UBE3A. En la mayoría de tejidos se expresan ambos alelos (sin impronta) pero en el cerebro solo se expresa el alelo materno (impronta específica de tejido). Es decir, mutaciones puntuales en la copia materna también son responsables del síndrome de Angelman.
Hay varios mecanismos genéticos que pueden inactivar o eliminar la copia materna del gen UBE3A:
- Deleción de un segmento del cromosoma materno 15 que contiene este gen.
- Mutación que inactiva la copia materna del gen UBE3A.
- Disomía uniparental paterna: herencia de dos copias del cromosoma 15 de su padre, en vez de una copia de la madre y una del padre.
- Translocación o por una mutación u otro defecto en la región del ADN que controla la activación del gen UBE3A. Estos cambios genéticos pueden anormalmente apagar UBE3A.
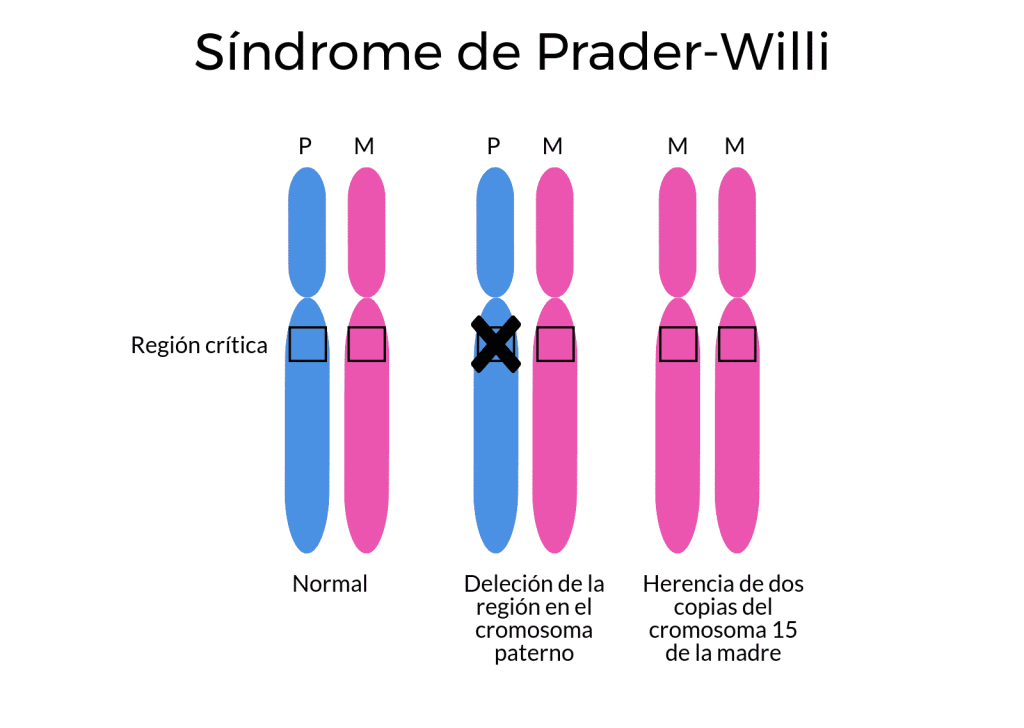
Ausencia de una copia de origen materno: síndrome de Prader-Willi
El síndrome de Prader-Willi se caracteriza por una sensación de hambre todo el tiempo acabando en obesidad, tono muscular pobre, capacidad mental reducida y órganos sexuales subdesarrollados. Está causado por la ausencia del transcrito SNURF-SNRPN, en la región 15, producido por el cromosoma paterno, junto con la deficiencia de algunos RNAs no codificantes en los intrones de SNURF-SNRPN. Como en el caso del síndrome de Angelman, el defecto puede ocurrir debido a que los genes del padre falten en el cromosoma 15, por problemas con los genes del padre en dicho cromosoma o bien, por la presencia de dos copias del cromosoma 15 de la madre y ninguna del padre.
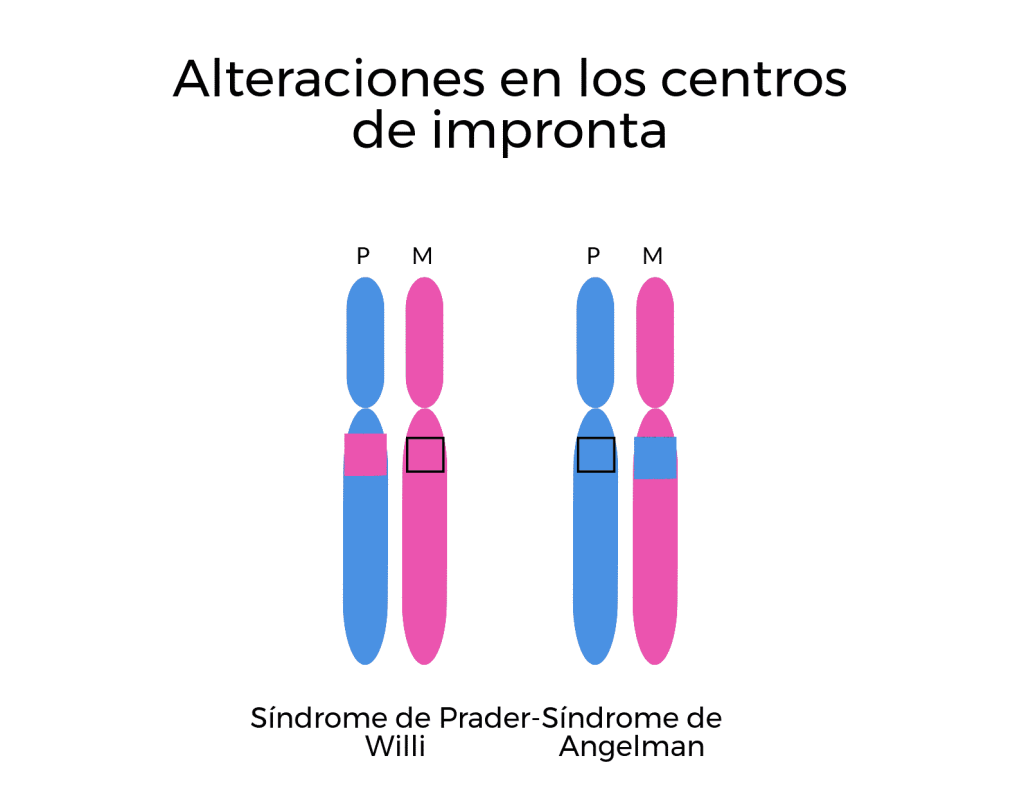
Pueden darse casos de algunos pacientes con el Síndrome de Prader-Willi y Angelman que tengan alteraciones en los centros de impronta. Implica que no se establezca de forma correcta la impronta durante la formación de los gametos de los pacientes. Se ha descrito un bajo número de pacientes que presentan mutaciones en los centros de impronta, se trata de deleciones que evitan que los espermatozoides y los óvulos adquieran la impronta que les corresponde. Por ejemplo, si un espermatozoide presenta una impronta femenina, los genes de Prader-Willi van a estar silenciados y, como el gameto femenino también los tiene silenciados, se dará el caso del síndrome de Prader-Willi. Si un óvulo es portador de impronta masculina, el gen UBE3A está silenciado en el gameto femenino y va a estarlo también en el gameto masculino. En este caso, por tanto, se dará el síndrome de Angelman.
Otras alteraciones relacionadas con el origen del material genético
Por último, otro problema relacionado con la impronta genómica gamética se ha detectado en algunos casos de niños nacidos a través de técnicas de reproducción asistida. Recientes estudios sugieren que hay mayor riesgo de enfermedades relacionadas con la impronta genómica, en niños nacidos mediante reproducción asistida, debido a que la conservación de los óvulos y los espermatozoides puede alterar la calidad de los mismos y, por tanto, alterar el establecimiento correcto de la impronta.
Por tanto, en situaciones como las descritas podemos observar la importancia de un correcto funcionamiento de los procesos genéticos en las primeras fases del desarrollo del zigoto. Podemos ver que no es suficiente con tener dos copias genéticas, sino que su origen es importante en el desarrollo de algunos tipos de patologías.
Bibliografía:
- Nussbaum, R.L, (2016), Genética en medicina, Barcelona, España, ELSEVIER.
- Juan Solari, A, (2004), Genética Humana. Fundamentos y aplicaciones en Medicina, Madrid, España, Editorial Panamericana.
- Jorde, L.B, (2010), Genética médica, Barcelona, España, ELSEVIER.
- Paoloni-Giacobino, A., Chaillet, R., (2004), Genomic imprinting and assisted reproduction. Reproductive Health, I (6), 1-7.
- Marques, C.J., Carvalho, F., Sousa, M., Barros. A., (2004), Genomic Imprinting in disruptive spermatogenesis, Lancet, 363, 1700-1702



