
Todos hemos estado dentro de un túnel.

De pronto, todo a tu alrededor se va desdibujando. Los laterales se vuelven borrosos y el foco de tu atención se va reduciendo, semejando la lente de una cámara que se cierra: el centro está enfocado pero todo lo que lo rodea está difuso, indefinido.
Así es la visión la visión de muchas personas con retinosis pigmentaria: ven la vida como en un túnel.
La retinosis pigmentaria no es una única enfermedad. Es el nombre que reciben un conjunto de enfermedades hereditarias raras caracterizadas por una pérdida lenta y progresiva de la visión. Esta patología afecta sobre todo a la retina, una capa delgada de tejido fotosensible encargada de la visión.
Cuando las células de la retina se deterioran, se experimentan síntomas como dificultad de visión nocturna y pérdida de visión periférica. En algunos casos avanzados, puede llegar a provocar la pérdida de visión.
La retinosis pigmentaria puede presentarse asociada a otras enfermedades (RP sindrómica), como ocurre en la enfermedad de Bassen-Kornzweig o el síndrome de Kearns-Sayre, o puede aparecer como enfermedad que afecta exclusivamente al ojo (RP no sindrómica), que es en la que nos centraremos.
Como su nombre indica, esta enfermedad afecta a la retina, por lo que primero vamos a ver qué es y cómo funciona la retina.
Qué es y cómo funciona la retina
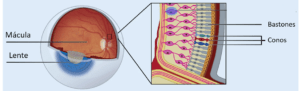
La retina es una capa delgada de tejido fotosensible del ojo, localizada en la parte posterior del mismo. Su función es transformar la luz que recibe en impulsos nerviosos, los cuales son enviados al cerebro a través del nervio óptico. Allí, se convertirán en las imágenes que percibimos.
La retina está constituida por dos capas: la interna o retina neural y la externa o epitelio pigmentario de la retina (EPR).
La retina neural está formada por varias capas de neuronas interconectadas entre sí, entre las que se encuentran los fotorreceptores (FR). En el ojo humano encontramos dos tipos de fotorreceptores: conos y bastones. Los conos se encuentran en la zona central y posterior de la retina. Se encargan de la visión en color y la nitidez de la visión central y se excitan (actúan) cuando hay luz. Por otra parte, los bastones se encuentran en la periferia de la retina. Sus funciones incluyen la visión en blanco y negro, visión periférica y visión nocturna. A diferencia de los conos, los bastones actúan en condiciones de poca luz.
Es importante entender la función de estos dos fotorreceptores, ya que tienen un patrón de degradación muy característico que ayudará con la diagnosis de la enfermedad.
Vista la estructura de esta capa, veamos ahora su funcionamiento.
La luz entra en el ojo atravesando la córnea, pupila, cristalino hasta llegar a la retina, donde las células fotorreceptoras reaccionan a esta luz excitándose, lo que provoca la liberación de sustancias químicas, que a su vez desencadenan un impulso nervioso.
Una vez realizada esta transformación de luz a señal eléctrica, la retina procesa la información que recibe y regula la que transmite al cerebro. La información se transporta codificada a través de la vía óptica hasta las áreas visuales cerebrales donde tiene lugar la percepción y comprensión visual.
Es decir, el patrón de luz que golpea las células fotorreceptoras desencadena un conjunto único de señales eléctricas, que el cerebro interpreta como una imagen.
La segunda capa de la retina es el epitelio pigmentario de la retina (EPR), una fina capa de células encargada de sostener y nutrir la retina. Concretamente, el EPR participa en el crecimiento y desarrollo del ojo, el mantenimiento de la retina y en el correcto funcionamiento de los fotorreceptores.
Origen de la retinosis pigmentaria: genes implicados
Como hemos definido al principio, la retinosis pigmentaria es una enfermedad hereditaria que afecta a la retina. Hasta el momento se han descrito más de 90 genes implicados el desarrollo de esta enfermedad, que en conjunto implican a cientos de mutaciones, que afectan a la función normal de la retina y pueden llevar a su degeneración progresiva.
Algunos de los genes implicados en el funcionamiento normal de la retina son:
- El gen RHO. Proporciona instrucciones para fabricar la proteína rodopsina, localizada en los bastones. Esta proteína está unida a una forma de vitamina A que cuando es golpeada por la luz, actica a la rodopsina. Esta activación desencadena una serie de reacciones químicas, que a su vez generan señales eléctricas que se transmitirán al cerebro. Finalmente en el cerebro, estas señales serán interpretadas como visión.
- El gen USH2A. Codifica para la proteína usherina, que se cree que forma parte de un complejo proteico con un papel clave en el desarrollo y mantenimiento de las células del oído interno y retina, además de en la sinapsis.
- El gen RPGR. Contiene la información necesaria para la síntesis de una proteína implicada en el movimiento celular, señalización química y percepción sensorial por cilios. Concretamente, se cree que una isoforma de esta proteína está especializada en el mantenimiento de los fotorreceptores a base de la regulación de los cilios.
- El gen CRB1. Codifica para una proteína involucrada en la determinación de la estructura y orientación de los fotorreceptores. La proteína CRB1 también puede estar implicada en la formación de conexiones entre diferentes tipos de células en la retina.
- Gen RP2. Sintetiza la proteína RP2, implicada en el transporte de proteínas dentro de los fotorreceptores.
Mutaciones en los genes implicados en el funcionamiento de la retinta pueden provocar la degeneración progresiva de conos y bastones siguiendo un patrón de pérdida de visión característico en personas con retinosis pigmentaria. Mutaciones en estos genes también pueden producir otras enfermedades que afectan a la visión.
Los bastones suelen estropearse antes que los conos, por lo que la visión nocturna deficiente suele ser el primer signo de la enfermedad. Más tarde, la visión diurna se ve afectada porque se pierden tanto los bastones como los conos.
Origen de la retinosis pigmentaria: herencia
Vistos algunos de los genes implicados, nos falta entender cómo se hereda la retinosis.
Aproximadamente el 50% de los casos de RP son aislados y no tienen antecedentes familiares.
Por otro lado, en los casos en los que sí se han establecido antecedentes familiares, se pueden distinguir tres categorías principales: autosómica recesiva, autosómica dominante y recesiva ligada al cromosoma X.
- Autosómica recesiva: ocurre cuando ninguno de los padres presenta la enfermedad pero ambos son portadores. Para que la enfermedad se manifieste, se debe heredar el gen recesivo (defectuoso) de ambos progenitores, siendo la probabilidad de que esto ocurra de ¼. El resto de la descendencia será portadora (2/4), lo que significa que no sufrirá retinosis pigmentaria, pero podrá transmitirla. Y por último, sólo ¼ de la descendencia estará completamente libre de la enfermedad. Mutaciones en el gen USH2A causan entre el 10-15% casos de RP en este tipo de herencia.
- Autosómica dominante: en este caso, una copia de un gen alterado en cada célula es suficiente para causar el trastorno. La mayoría de las personas con retinosis pigmentaria autosómica dominante tienen un progenitor afectado y otros miembros de la familia con el trastorno. En este caso, la probabilidad de que la descendencia se vea afectada por la enfermedad es de una entre dos cuando un progenitor tiene un gen normal y uno Este es el tipo de herencia asociado a las mutaciones en el gen RHO.
- Recesiva ligada al cromosoma X: siendo el padre enfermo o siendo la madre portadora. En las familias con el herencia recesiva ligada al cromosoma X, solo los hombres se ven afectados, mientras que las mujeres portan el rasgo genético pero no experimentan una pérdida severa de la visión. Si el padre tiene retinosis, ningún hijo será enfermo y todas las hijas serán portadoras. Si la madre es la portadora, 1 de cada 2 hijos se ve afectado y 1 de cada 2 hijas es portadora. Las mutaciones en el gen RP2 causan este tipo de herencia.
Síntomas de la retinosis pigmentaria
Debido a su heterogeneidad genética los síntomas pueden variar entre pacientes. Los más comunes son:
- Ceguera nocturna. Como su nombre implica, la ceguera nocturna es cuando no se ve nada en la oscuridad. Ocurre como consecuencia de la disfunción de los bastones. Es el síntoma más precoz y puede ser la única manifestación de la enfermedad durante años.
- Lenta adaptación a la oscuridad. El tiempo de adaptación de la visión al pasar de un ambiente bien iluminado a otro peor iluminado aumenta mucho. Es decir, la visión tarda mucho en adaptarse a las nuevas condiciones de iluminación. Esto ocurre como consecuencia de la degradación de conos, que se da cuando la retinosis es más avanzada.
- Reducción progresiva del campo visual. Consiste en la pérdida de la visión periférica, dificultando la localización de los objetos que nos rodean. Esto ocurre, como hemos visto antes, por el deterioro de los bastones, lo que provoca que el campo visual se vaya estrechando progresivamente, quedando en fases avanzadas lo que se conoce como visión en túnel.
- Disminución de la visión. Es el último síntoma en aparecer y es un signo de lo avanzada que está la enfermedad. Puede incluso alterar la percepción de los colores.
También es común que las personas con retinosis pigmentaria presenten cataratas a temprana edad además de inflamación de la retina.
Diagnóstico
Existen diversas formas de diagnosticar la RP:
- Electrorretinograma (ERG). Este test mide la actividad eléctrica de los fotorreceptores, que será baja en personas con RP.
- Prueba de campo visual. Esta prueba determina cuánta visión se ha perdido, generando un mapa del campo visual del paciente La persona ha de pulsar un botón para indicar cuándo observa una luz que irá desplazándose 180 grados.
- Pruebas genéticas. Este tipo de pruebas puede identificar la causa genética exacta, información que puede facilitar hacer predicciones sobre la evolución o tomar decisiones sobre el diagnóstico. Es la forma más precisa de diagnóstico.
Tratamiento
Se están realizando ensayos clínicos para evaluar nuevos tratamientos para la retinosis pigmentaria. Entre ellos encontramos el uso de DHA, un ácido graso omega-3 en desarrollo. También se está evaluando el tratamiento con antioxidantes, como el palmitato de vitamina A, que en dosis elevadas podría retrasar la aparición de la enfermedad. Por otra parte, tomar altas dosis de vitamina A puede causar problemas hepáticos graves, por lo que los beneficios del tratamiento deben sopesarse frente a los riesgos para el hígado.
El foco de investigación actual se encuentra en la terapia genética y en los trasplantes y prótesis de retina. Como la retinosis resulta, generalmente, de un gen defectuoso, la terapia génica se ha convertido en la principal herramienta para la lucha contra esta enfermedad. El objetivo de las investigaciones reside en introducir «genes sanos» en la retina para favorecer su correcto funcionamiento.
Concluyendo, a pesar de haber investigaciones prometedoras, los resultados todavía no se consideran ni seguros ni exitosos. Lo que sí encontramos son recomendaciones como el uso de gafas de sol para proteger la retina de la luz ultravioleta o revisiones frecuentes con el oftalmólogo para la detección rápida de cataratas o inflamación de la retina, ambos problemas que se pueden abordar y pueden aparecer como consecuencia de la retinosis.
Fuentes y enlaces de interés
MedlinePlus. https://medlineplus.gov/spanish/
National eye institute. https://www.nei.nih.gov/
Qué es la retina. https://www.oftalmoseo.com/patologias-frecuentes-2/que-es-la-retina/
https://www.topdoctors.es/diccionario-medico/retina#
National Insitute of Biotechnology Information. https://www.ncbi.nlm.nih.gov/
Federación Española de Enfermedades Raras. https://enfermedades-raras.org/index.php?option=com_content&view=article&id=973&Itemid=171
Retinosis retina begisare: https://www.begisare.org/
Funcionamiento de la retina. https://www.clinicasnovovision.com/blog/funcionamiento-de-la-retina/
Institut català de la retina. https://icrcat.com/
Sociedad Española de Oftanmología. https://www.oftalmoseo.com/patologias-frecuentes-2/que-es-la-retina/
Orphanet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=480
OMIM: https://omim.org/phenotypicSeries/PS268000
GeneReviews: https://www.ncbi.nlm.nih.gov/books/NBK1417/
Si te ha gustado este blog y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria



