
INTRODUCCIÓN
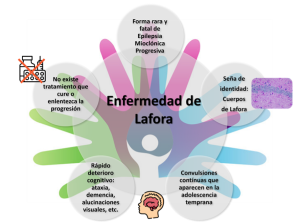
La epilepsia mioclónica progresiva de tipo Lafora —o sencillamente enfermedad de Lafora— es una enfermedad de muy baja prevalencia, clasificada por tanto como enfermedad rara (ORPHA:501). Se trata de una gravísima condición, de carácter incurable y altamente incapacitante, que se manifiesta en la adolescencia y con un desarrollo dramáticamente veloz (Markussen et al., 2021). Su base es genética, de carácter recesivo (OMIM: 254780) y si bien durante las últimas décadas se ha avanzado de manera asombrosa en la comprensión de su fisiopatología, todavía no se dispone de un tratamiento efectivo que aumente la calidad de vida de los pacientes y contribuya a mejorar su pronóstico. No obstante, el complejo panorama molecular descrito en los últimos años ha terminado por aportar importantes pistas con un potencial para el desarrollo de nuevas terapias que está empezando a explotarse. Es por tanto objeto de la presente revisión hacer, por un lado, un repaso histórico de la enfermedad (destacando los hallazgos de mayor relevancia a nivel tanto clínico como de investigación) y por otro, revisar los últimos avances capaces de contribuir al desarrollo de nuevas estrategias terapéuticas, para reflexionar sobre el futuro de estas investigaciones y las implicaciones de las mismas para la vida de los pacientes y sus familiares.
En primer lugar, presentaremos esta particular enfermedad retrotrayéndonos a la primera persona que la describió, y de quien heredó su nombre.
Lafora, discípulo y maestro
Gonzalo Rodríguez Lafora (1886–1971) fue uno de los más significativos de entre los muchos discípulos que legaría Santiago Ramón y Cajal. Formado como neuropsiquiatra, llegó a publicar más de un centenar de trabajos que abarcan desde tratados de histopatología hasta artículos de investigación básica. Director del Laboratorio de Fisiología Cerebral en la Junta para Ampliación de Estudios (JAE) durante la primera etapa de la misma (en la que el propio Cajal ejercía de presidente), fue a su vez maestro de muchos científicos y científicas: bajo sus órdenes trabajó Soledad Ruiz-Capillas, primera española con una carrera universitaria que entraba en un laboratorio como el de Cajal y pionera de la neurociencia en nuestro país (Giné et al., 2019). Pese a sus indudables logros en el campo de la neuropsiquiatría y en lo tocante al desarrollo de la investigación científica en nuestro país (Nanduri et al., 2008), Lafora es recordado internacionalmente por su descripción pionera de la patología que adquiriría su nombre y ocupa el tema central de esta revisión: la epilepsia mioclónica progresiva de tipo Lafora.
Esta terrible enfermedad neurodegenerativa fue descrita por Lafora durante su estancia en el Hospital for the Insane en Washington, D.C. entre 1909 y 1912. Lafora venía ya entonces de haberse formado en Alemania y Francia, habiendo trabajado con el mismo Alois Alzheimer, y su conocimiento de las técnicas histopatológicas junto a su habilidad para ilustrar los hallazgos observados bajo el microscopio (sin duda herencia de su formación con Cajal) propiciaron que los primeros artículos donde se describe el primer caso registrado de esta rara epilepsia, publicados ambos en 1911, constituyan documentos de gran valor científico. En el primero de ellos (Lafora, 1911), Lafora describe con profusión el rasgo más característico de la enfermedad y que sigue centrando la mayor parte de atención incluso en el panorama investigador actual: las inclusiones intracelulares presentes en los cuerpos neuronales, que han dado en llamarse cuerpos de Lafora y que el propio científico describió como «cuerpos amiloides». En el artículo con Bernhard Glueck como coautor (Lafora y Glueck, 1911) y titulado Beitrag zur Histopathologie der myoklonischen Epilepsie (Contribución a la histopatología de las epilepsias mioclónicas) describe las características clínicas de un paciente de 17 años cuya autopsia daría lugar a las mencionadas observaciones histopatológicas, y que constituyen a fecha de hoy el paradigma de la sintomatología de la enfermedad: mioclonías constantes, demencia progresiva, pérdida de capacidad auditiva y visual, y muerte por status epilepticus.
Desde esta sucinta descripción habrían de pasar noventa años hasta que la enfermedad de Lafora consolidase su definición (Minassian, 2001) y se esclareciese su base genética. Durante ese periodo algunos síntomas se fueron añadiendo a la ya de por sí dilatada lista (afasia, ataxia, ausencias, crisis tónico-clónicas,…) y se constató el carácter hereditario recesivo, causa de la muy baja prevalencia de la enfermedad (hoy en día se estima entre 1-9 por cada millón según Orphanet). Muchos de los síntomas y especialmente el debut durante la infancia tardía o adolescencia podrían presentar confusión frente a otro tipo de epilepsias o trastornos neurológicos, pero desde los primeros trabajos de Lafora, el diagnóstico diferencial consistió en la identificación de los cuerpos intracelulares que, además de en neuronas, se acumulan en mayor cantidad en otro tipo celular del cerebro, los astrocitos. Los cuerpos de Lafora se acumulan, además de en cerebro, en otros tejidos periféricos como hígado, músculo esquelético, corazón o piel, siendo la biopsia de este último tejido el método más extendido para realizar dicho diagnóstico diferencial (Busard et al., 1991) (Figura 1). El descubrimiento de que dichos cuerpos estaban constituidos por poliglucosanos, y por tanto fácilmente identificables en cortes histológicos por tinción mediante PAS o técnica de Shiff (basada en ácido peryódico) (Figura 1) hizo de la biopsia de piel el método definitivo para diagnosticar la enfermedad de Lafora hasta el descubrimiento de las bases genéticas de la enfermedad. Actualmente la estandarización de los métodos de secuenciación y diagnóstico genético han permitido analizar un amplio abanico de mutaciones en los pacientes de Lafora, como veremos a continuación.
Dos genes, una única enfermedad
El primer gen directamente relacionado con este tipo de epilepsia mioclónica fue identificado a finales de la década de los 1990 (Minassian et al., 1998; M.Serratosa et al., 1995; Serratosa et al., 1999) y denominado EPM2; más adelante se renombraría como EPM2A tras la identificación de un segundo locus que también apareció asociado a otros pacientes aquejados de la epilepsia de Lafora, y que se etiquetaría como EPM2B/NHLRC1 (Chan et al., 2003) (Figura 2). Curiosamente, ambos genes están localizados en diferentes regiones del cromosoma 6 (EPM2A en 6q24.3 y 6p22.3 en el caso de EPM2B) pero codifican para proteínas totalmente distintas a nivel estructural y funcional, y se ha llegado a describir hasta 80 mutaciones relacionadas con la enfermedad para cada gen (http://projects.tcag.ca/lafora). Durante las últimas décadas la evidencia acumulada en cuanto a las propiedades del producto proteico de cada uno de los genes ha supuesto un avance muy significativo en la comprensión de las bases moleculares de la patología, como se detallará en el siguiente apartado; pero antes describiremos sucintamente ambas proteínas.
El locus EPM2A codifica para una proteína que se denominaría laforina, perteneciente en base a su secuencia de aminoácidos y estructura molecular a la superfamilia de fosfatasas de tirosina, y en concreto a la subfamilia de fosfatasas de especificidad dual (Dual Specificity Phosphatases, DSP) (Serratosa et al., 1999) (Figura 2). Dentro de este grupo se clasifican muchas proteínas con actividad catalítica de tipo fosfatasa, basada en un aminoácido de cisteína y susceptibles a inhibición por vanadato sódico. La mayoría son capaces de eliminar grupos fosfato en aminoácidos de serina y treonina. Pero, además, pueden desfosforilar otros sustratos no proteicos (tal es el caso del famoso supresor tumoral PTEN con especificidad hacia fosfatidil inositol trifosfato, un fosfolípido con actividad señalizadora). Laforina presenta la particularidad de ser la única proteína de este gran grupo, y de hecho la única codificada por el genoma humano, capaz de desfosforilar carbohidratos complejos y más en concreto, el glucógeno. Esta propiedad fue descrita poco tiempo después de la identificación del locus al definir que el módulo catalítico característico de la familia de DSP estaba precedido en la secuencia de laforina por un módulo de unión a carbohidratos (CBD, carbohydrate-binding domain) (Wang et al., 2002), aunque se tardaría casi veinte años en resolver la estructura completa de la proteína para poder tener una visión clara de cómo ambos módulos se organizan tridimensionalmente (Raththagala et al., 2015) (Figura 2). La descripción de las propiedades bioquímicas de laforina fue clave para comprender mejor la naturaleza de las inclusiones conocidas como cuerpos de Lafora, constituidas principalmente por la acumulación de una forma aberrante de glucógeno, hiperfosforilado y menos ramificado que su contrapartida fisiológica, lo que redunda en una menor solubilidad y la tendencia a formar agregados y precipitados intracelulares (Sakai et al., 1970). Más adelante se definiría que dichas inclusiones contenían también ubicuitina (Ganesh, 2002) y proteínas relacionadas con la regulación de la proteostasis celular (Rao et al., 2010), lo que encaja perfectamente con la función biológica no de laforina, sino de malina, la otra proteína codificada por el locus EPM2B.
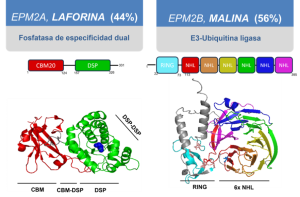
Malina (del término francés para definir la epilepsia, grand mal) es la proteína codificada en este segundo locus y se caracteriza por la concatenación de fragmentos de secuencia con el dominio NHL —que tridimensionalmente conducen a la formación de una estructura «en barril»— precedidos por un dominio RING, lo cual le propició el nombre alternativo de NHLRC1 (NHL repeat containing 1) (Chan et al., 2003) (Figura 2). A nivel bioquímico, la actividad catalítica de esta enzima es de tipo E3 ubicuitina ligasa. Estas proteínas participan en el último paso en el proceso de ubicuitinación de proteínas, mediante el cual la adición de una cadena de poli-ubicuitinas sirve para etiquetar cualquier diana para facilitar su interacción con maquinaria celular responsable de cambiar su función o, muy habitualmente, producir su degradación a través de diferentes vías como pueden ser la del proteasoma o la vía autofágica. Las enzimas E3 facilitan la interacción entre las enzimas conjugadoras de ubicuitina (tipo E2) y los sustratos finales, promoviendo la adición de la cadena de poli-ubicuitinas sobre la proteína diana (Kerscher et al., 2006; Li y Ye, 2008; Ye y Rape, 2009).
Recapitulando este panorama genético y las proteínas correspondientes, nos encontramos con defectos de función en una proteína fosfatasa de carbohidratos o en una proteína capaz de degradar sustratos; y en ambos casos, dichos defectos redundan en la acumulación de formas alteradas de glucógeno que terminan por ocupar el citoplasma celular de forma patológica. La relación funcional definitiva entre ambas proteínas, más allá de que puedan participar en promover la aparición de los cuerpos de Lafora, se terminó de perfilar cuando se evidenció que ambas proteínas forman un complejo funcional mediante interacción directa (Gentry et al., 2005) y, a su vez, dicho complejo interaccionaba físicamente con otras proteínas reguladoras de la formación y/o eliminación del glucógeno (Kumarasinghe et al., 2023; Moreno et al., 2010; Romá-Mateo et al., 2011; Rubio-Villena et al., 2013). Las piezas clave para la comprensión de la enfermedad de Lafora estaban, por tanto, y casi cien años después de su descripción inicial, puestas sobre la mesa. No obstante, durante los últimos diez años este puzle ha ido enriqueciéndose con multitud de datos que definen aún mejor las consecuencias fisiopatológicas de las alteraciones moleculares que apenas acabamos de describir, lo cual ha conducido a un periodo en el que, por vez primera, se comienza a perfilar nuevos horizontes y estrategias terapéuticas centradas en la fina disección de vías moleculares tan complejas como las que seguiremos describiendo a continuación.
BASES FISIOPATOLÓGICAS Y MOLECULARES: PEQUEÑOS ORGANISMOS, GRANDES HALLAZGOS
Para profundizar en el conocimiento de la enfermedad de Lafora, ha sido necesario crear modelos que permitieran avanzar en la comprensión de la enfermedad y a su vez desarrollar posibles terapias. A continuación, presentaremos los principales modelos animales y celulares empleados para el estudio de la enfermedad, para más adelante profundizar en las alteraciones celulares y moleculares subyacentes a la enfermedad que nos han permitido conocer con un alto grado de detalle.
Modelos animales y celulares
La mayoría de hallazgos sobre la fisiopatología de la enfermedad se han obtenido, como suele ser habitual en la investigación de enfermedades genéticas durante las últimas décadas, a partir de modelos experimentales de ratón; no obstante, existen otros animales que han contribuido a avanzar en nuestro conocimiento de la enfermedad.
- Pez cebra
Recientemente se ha obtenido un modelo de la enfermedad en el pez cebra para investigar la pérdida de función de laforina (Della Vecchia, et al., 2022; Della Vecchia, Ogi, et al., 2022). Este modelo, en el que se ha eliminado por completo la expresión del gen (lo que se abrevia como Epm2a-/-) presenta las principales características de la enfermedad tales como: deficiencia motora, hiperexcitabilidad neuronal con crisis epilépticas espontáneas, aumento de la respuesta inflamatoria y alteración de la autofagia. Además de la caracterización de este nuevo modelo animal, Della Vechia va un paso más allá e identifica que la administración precoz de trehalosa mejora la actividad motora y reduce la hiperexcitabilidad neuronal asociada a las crisis. La ventaja de este modelo frente a los modelos mamíferos, es que se trata de una nueva herramienta más sencilla para investigar nuevos tratamientos ya que presenta menos restricciones normativas y es mucho más económico.
- Perro
En perros, la enfermedad puede aparecer espontáneamente en cualquier raza, pero afecta especialmente a los llamados perros salchicha, los basset hounds y los beagles (Gredal et al., 2003; Kaiser et al., 1991; Lohi, Young, et al., 2005). A diferencia de los humanos, en perros está causada por una mutación de expansión repetida en el gen EPM2B donde los individuos afectados son portadores de 19 a 26 copias de la secuencia repetida en lugar de las 2-3 copias en condiciones normales (Hajek et al., 2016; Lohi, Young, et al., 2005; von Klopmann et al., 2021). La enfermedad aparece a partir de los 5 años de edad y los animales afectados tienen una esperanza de vida casi normal. La principal característica de la enfermedad de Lafora canina es que la mioclonía puede ser inducida por luces intermitentes, sonidos repentinos y movimiento, y a medida que progresa van apareciendo otros trastornos neurológicos como ataxia, ceguera y demencia. Al igual que en los humanos, los cuerpos de Lafora se distribuyen en el cerebro, hígado y corazón y son idénticos en estructura y tamaño (Lohi, Young, et al., 2005). En cerebro, estos cuerpos se encuentran principalmente en astrocitos y neuronas, y también hay astrogliosis y microgliosis reactiva (Chambers et al., 2018; Hegreberg y Padgett, 1976; Menchetti et al., 2021).
- Ratón
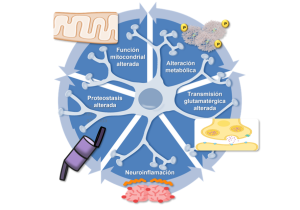
Como hemos comentado, este es el modelo de enfermedad del que se dispone de más estudios y por tanto es el mejor caracterizado. Tras la identificación de los dos genes Epm2a y Epm2b como responsables de la enfermedad, se han generado varios modelos de ratón mediante la interrupción de ambos genes (Chan, 2004; DePaoli-Roach et al., 2010; Ganesh, 2002) (Criado et al., 2012), lo que se conoce como modelos knock-out (KO); en este apartado nos centraremos en los KO Epm2a-/- (Ganesh, 2002) y Epm2b-/- (Criado et al., 2012). Estos ratones presentan cuerpos de Lafora y complicaciones neurológicas que se asemejan a las presentes en los pacientes. Tanto los ratones Epm2a-/- como los Epm2b-/- presentan déficits de memoria, de actividad motora y coordinación, ataxia, debilidad muscular, discinesias, diferentes niveles de actividad epiléptica espontánea y mayor susceptibilidad al quimioconvulsivante PTZ (pentilenotetrazol) (Criado et al., 2012; Ganesh, 2002; García-Cabrero et al., 2014; García-Cabrero et al., 2012). Estos defectos funcionales correlacionan con muerte neuronal, aumento del estrés oxidativo, así como con proteostasis y autofagia alterada (Aguado et al., 2010a; D. Burgos et al., 2020; Knecht et al., 2010; Lahuerta et al., 2018; Muñoz-Ballester et al., 2016, 2019; Puri et al., 2012; Rao et al., 2010; Romá-Mateo, Aguado, García-Giménez, Ibáñez-Cabellos, et al., 2015b; Vernia et al., 2009) (Figura 3). Asimismo, se ha podido determinar que la neuroinflamación es una característica singular de la enfermedad en los ratones, principalmente desencadenada por los mediadores TNF e IL-6 y con los marcadores epigenéticos mirR-146a y miR-155 aumentados (Lahuerta et al., 2018; Romá-Mateo et al., 2023a; Rubio et al., 2023) (Figura 3). Además, la presencia de cuerpos de Lafora en el cerebro se ha asociado con las anomalías neurológicas reportadas en los ratones Epm2a-/- y Epm2b-/-, lo que sugiere que estos agregados podrían causar deterioro cognitivo y conductual (Criado et al., 2012; García-Cabrero et al., 2012). Por último, comentar que se han evaluado varios tratamientos farmacológicos en estos modelos murinos que se tratarán con más detalle en la sección “Nuevas aproximaciones terapéuticas”.
Modelos celulares
El trabajo con líneas celulares es fundamental para la investigación biomédica. En el caso de enfermedades de tan baja prevalencia, es muy difícil disponer de modelos celulares derivados de pacientes, y lo más habitual es utilizar líneas modelo que se utilizan genéricamente para experimentos de biología celular y molecular. No obstante, a partir de los modelos animales se pueden obtener cultivos de células que permiten otro tipo distinto de aproximaciones especialmente útiles en el laboratorio: en el caso de la enfermedad de Lafora, se han empleado varios modelos celulares procedentes de modelos murinos. A partir de fibroblastos embrionarios de ratones Emp2a-/- y Emp2b-/-, así como en la línea celular de neuroblastoma Neuro2a, en la que se ha silenciado parcialmente la expresión de Epm2a y Epm2b, se ha podido determinar una alteración de la proteostasis y una reducción del flujo autofágico (Aguado et al., 2010a; Criado et al., 2012; Jain et al., 2017) (Garyali et al., 2014a). Además, recientemente hemos descrito que los cultivos de astrocitos de ratones Epm2a-/- y Epm2b-/- acumulan más glucógeno —probablemente como consecuencia de una mayor captación de glucosa— y validamos su uso como plataforma in vitro para realizar escrutinio de fármacos que pueda identificar nuevos tratamientos para la enfermedad (Moreno-Estellés et al., 2023). Por otra parte, la disminución de la expresión de EPM2A en la línea celular renal de mono COS7 ha permitido determinar que la inhibición del gen SGK1 podría ser una estrategia terapéutica para evitar la acumulación de glucógeno (Singh et al., 2013).
Por último, destacar que, aun siendo muy difícil su obtención, sí se ha llegado a emplear fibroblastos de pacientes de enfermedad de Lafora con mutaciones en el gen EPM2A que han permitido identificar la importancia de Laforina en la autofagia y su regulación a través de la ruta de la rapamicina quinasa dependiente/mTOR (Aguado et al., 2010b).
La síntesis de glucógeno, primera gran alteración celular
Las células almacenan glucosa en forma de una molécula compleja llamada glucógeno, a modo de reservorio energético. En la síntesis de glucógeno participan dos enzimas de forma coordinada: por una parte, la enzima glucógeno sintasa (GS, del inglés Glycogen Synthase) la cual une glucosas entre sí mediante enlaces glicosílicos α-1,4; y la enzima glucógeno ramificante (GBE, Glycogen branching enzyme), la cual crea puntos de ramificación a través de enlaces α-1,6. De forma equivalente, en la degradación de glucógeno intervienen dos enzimas, la glucógeno fosforilasa (GP, Glycogen phosphorylase) y la enzima desramificante (GDE, Glycogen debranching enzyme) (Adeva-Andany et al., 2016). Una molécula de glucógeno puede llegar a almacenar hasta 55.000 unidades de glucosa a través de ramificaciones estructuradas generando una esfera soluble que puede degradarse rápidamente cuando la célula necesita glucosa. En el caso de los cuerpos de Lafora, el glucógeno está poco ramificado, y las cadenas largas de glucosa se agregan formando hélices simples o dobles que excluyen el agua impidiendo su degradación. Otra particularidad del glucógeno en los cuerpos de Lafora es que está hiperfosforilado contribuyendo de nuevo a su insolubilidad (Roach, 2015). Este glucógeno anómalo, al no ser accesible a las enzimas responsables de su movilización (GP y GDE), no se degradada y por ello empieza a acumularse. Además del glucógeno aberrante, los cuerpos de Lafora alojan en su composición proteínas tales como la GS, subunidades del proteasoma, componentes de la ruta autofágica, proteínas ubicuitinadas, y proteínas de choque térmico, entre otras (Duran et al., 2014; Pellegrini et al., 2022; Varea et al., 2021). Durante años, la investigación en enfermedad de Lafora ha estado centrada en averiguar todos los detalles de estos omnipresentes cuerpos, que como agujeros negros celulares atraen y aglutinan en su interior a componentes que son críticos para la vida celular, alterando su fisiología; ya el propio Lafora se preguntaba si serían causa o consecuencia de la enfermedad, y a fecha de hoy sigue abierto un interesante debate en cuanto a si son los causantes de la muerte neuronal o más bien un silencioso testigo, precisamente, de los esfuerzos por las neuronas por sobrevivir a los defectos producidos por la falta o mal funcionamiento de laforina o malina.
Como ya se ha mencionado en el apartado anterior, la epilepsia de Lafora es una enfermedad autosómica recesiva causada por mutaciones en dos genes: EPM2A, que codifica para laforina, enzima que elimina grupos fosfato de carbohidratos (Minassian et al., 1998; Serratosa et al., 1999), y EPM2B/NHLRC1, que codifica para malina, una E3-ubicuitina ligasa que une ubicuitinas a otras moléculas (Chan et al., 2003). Pacientes con mutaciones en laforina o en malina son indistinguibles, y esto se debe a que laforina y malina forma un complejo funcional (Gentry et al., 2005; Lohi, Ianzano, et al., 2005; Solaz-Fuster et al., 2008), y cuando uno de los dos componentes falla, el complejo pasa a ser disfuncional. Aunque no se conoce la función exacta de malina y laforina en el metabolismo del glucógeno, está ampliamente aceptado que laforina recluta a malina para unirse al glucógeno y ubicuitinar a diferentes proteínas implicadas en dicho metabolismo (Cheng et al., 2007; Solaz-Fuster et al., 2008; Worby et al., 2008).
A continuación, se presentarán algunos de los principales sustratos del complejo laforina-malina implicados en el metabolismo del glucógeno:
- GS y GDE
La enzima glucógeno sintasa GS, responsable de la síntesis de glucógeno (Vilchez et al., 2007), y la enzima desramificante GDE, implicada en la degradación de glucógeno, son sustratos directos del complejo (Cheng et al., 2007). Así, cuando el complejo no funciona, aumentan los niveles de estas dos enzimas y esta situación se traduce en un aumento de la cantidad de glucógeno poco ramificado.
- PPP1R3C
PPP1R3C o también conocida como R5/PTG, es una subunidad reguladora de la fosfatasa PP1, la cual es responsable de la activación de la enzima GS (síntesis de glucógeno) y la inactivación de la enzima GP (degradación del glucógeno). En condiciones fisiológicas, el complejo laforina-malina ubicuitina a PPP1R3C promoviendo su degradación. Así, la ausencia de PPP1R3C supone la inactivación de la síntesis y la activación de la degradación glucógeno. Sin embargo, en el caso de la enfermedad de Lafora, al no ser funcional el complejo laforina-malina, la presencia de PPP1R3C promueve la síntesis de glucógeno y frena su degradación (Solaz-Fuster et al., 2008; Vilchez et al., 2007; Worby et al., 2008). Otras subunidades reguladoras de la fosfatasa PP1, tales como PPP1R3B (Worby et al., 2008) y PPP1R3D (Rubio-Villena et al., 2013), también han sido identificadas como sustrato del complejo laforina-malina.
Defectos en autofagia y proteostasis: la otra cara de los cuerpos de Lafora
La descripción detallada de la composición de los cuerpos de Lafora (Puri et al., 2011), junto a las numerosas evidencias en cuanto al rol del complejo laforina-malina en la degradación de proteínas específicas y los mecanismos por los que esta se lleva a cabo, condujo a la definición de una serie de alteraciones a nivel celular que completaron el panorama —ya de por sí complejo— de la fisiopatología de la enfermedad a nivel intracelular (Gentry et al., 2013; Romá-Mateo et al., 2012) (Figura 3). Los mecanismos de señalización mediante ubicuitina presentan una gran complejidad, e incluyen una amplia diversidad de modificaciones asentada en la capacidad de generar cadenas de poliubicuitina no solo de diferente longitud (mono/poliubicuitina) sino de diferente topología, mediante la alternancia entre cadenas ramificadas que parten de la polimerización de monómeros de ubicuitina a partir de diferentes lisinas iniciales de la propia molécula. Las más estudiadas son la polimerización a través de la lisina 48 y la de la lisina 63, relacionadas con degradación a través del proteasoma en el primer caso y de la autofagia en el segundo (Ikeda y Dikic, 2008). Experimentalmente, utilizando los modelos celulares y animales disponibles como modelo de enfermedad de Lafora, se ha podido definir el tipo de ubicuitinación mediado por malina en cada uno de los casos en que se ha descrito una diana específica, comprobándose que ambos tipos se producen. En el caso de la degradación mediada por autofagia, adicionalmente, se ha definido la implicación de una enzima de tipo E2 específica —la proteína UBE2N— y la mediación adicional del regulador de autofagia p62 (también llamado sequestosome-1) (Sánchez-Martín et al., 2015). Cabe mencionar, no obstante, que el concepto de autofagia en sentido amplio incluye multitud de procesos específicos, clasificados en torno al tipo de estructuras cuya degradación es mediada en cada caso. La disección de los reguladores implicados en autofagia alterados en enfermedad de Lafora ha permitido descubrir, por ejemplo, importantes defectos en el procesamiento de mitocondrias dañadas (proceso conocido como mitofagia) (Lahuerta et al., 2018). Estos resultados dan soporte a las observaciones realizadas con anterioridad referentes a desequilibrios en parámetros de estrés oxidativo e inflamación que tienen una relación directa con los procesos que acabamos de mencionar, como veremos a continuación y describiremos en detalle en la sección siguiente.
Este tipo de trabajos han permitido definir de una manera muy completa el panorama molecular de las alteraciones debidas a fallos en cualquiera de las dos proteínas implicadas, y han sido confirmadas por el hallazgo de multitud de alteraciones en el funcionamiento tanto de la actividad proteasomal como de la vía de la autofagia y los procesos que esta regula en los modelos celulares y animales de la enfermedad (Aguado et al., 2010a; Garyali et al., 2014; Jain et al., 2017; Knecht et al., 2012). Es interesante que estos hallazgos suelen ser prácticamente equivalentes tanto en modelos deficientes para el gen EPM2A como EPM2B. Además, al centrar la atención en la alteración de estos mecanismos de mantenimiento de la homeostasis a nivel de estructuras proteicas (lo que llamamos proteostasis) se ha puesto el foco en otros mecanismos de transducción de señales mediados por moléculas y complejos cuya alteración da lugar a un incremento en estrés oxidativo e inflamación. Ambos procesos están ampliamente relacionados y son fuente de numerosas estrategias basadas tanto en diseño de biomarcadores como en estrategias terapéuticas, por lo que merece la pena detenerse a definirlos en mayor profundidad.
Estrés oxidativo e inflamación, o la pescadilla que se muerde la cola
El estrés oxidativo —definido como el desequilibrio entre la producción de especies reactivas de oxígeno y la respuesta antioxidante celular encargada de regularla— constituye uno de los factores que más contribuyen a la fisiopatología de múltiples enfermedades. Las especies reactivas de oxígeno y nitrógeno forman parte de los procesos de señalización intracelular fisiológicos, pero su producción excesiva y descontrolada da lugar a alteraciones no solo en dichas vías, sino a modificaciones que pueden llegar a ser irreversibles en componentes celulares como ácidos nucleicos, proteínas y fosfolípidos. Es difícil, a menudo, discernir entre el estrés oxidativo generado como fenómeno inicial en base a defectos genéticos que afectan directamente a los reguladores de la respuesta antioxidante y el generado como consecuencia de otras alteraciones celulares. Por ejemplo, y de gran relevancia para el caso de la enfermedad que nos ocupa, una respuesta deficiente en los mecanismos de eliminación de estructuras proteicas dañadas o en la gestión de la eliminación de mitocondrias defectuosas contribuye en gran medida a incrementar el estrés oxidativo celular. Las especies reactivas producidas en exceso reaccionan con las moléculas circundantes, y a su vez pueden dañar tanto a los componentes del proteasoma como a los intermediarios de las vías de la autofagia (Wu et al., 2009).
Por lo tanto, y dados los antecedentes descritos para el complejo panorama molecular en la epilepsia de Lafora, no es de extrañar que el estrés oxidativo haya sido descrito como parte de las alteraciones encontradas en modelos celulares y animales de la enfermedad (Figura 3). Nuestros grupos de investigación han profundizado en este aspecto de la patología, dando lugar a interesantes hallazgos (Romá-Mateo, Aguado, García-Giménez, Ibáñez-Cabellos, et al., 2015a). Entre los más relevantes a este respecto encontramos una disfunción mitocondrial acompañada de una producción exacerbada de ion superóxido en líneas celulares de fibroblastos humanos con mutaciones tanto en EPM2A como en EPM2B, concomitante a una desregulación de la actividad antioxidante mediada por las enzimas superóxido dismutasa mitocondrial (MnSOD), citosólica (CuZnSOD) y catalasa. Estas alteraciones fueron confirmadas en ambos modelos de ratón, Epm2a-/- y Epm2b-/- en los que también se halló una expresión alterada de la proteína peroxirredoxina 6 (Prdx6) dependiente de glutatión y relacionada con la peroxidación lipídica (Romá-Mateo, Aguado, García-Giménez, Ibáñez-Cabellos, et al., 2015) (Fisher et al., 2018). Estos interesantes resultados demostraron además una importante relación entre los procesos de respuesta antioxidante y la regulación de la proteostasis celular: algunas de las alteraciones mencionadas se vieron exacerbadas al cultivar las células en condiciones de deprivación de nutrientes, las cuales se utilizan para estimular las vías proteolíticas. Esta relación entre degradación de estructuras celulares o proteínas y la respuesta antioxidante se vio respaldada por un trabajo previo, donde habíamos demostrado que la proteína de respuesta antioxidante tiorredoxina 1 (Trx1) y la subunidad 20S del proteasoma interaccionaban físicamente; interacción que se veía afectada tanto como la función de ambos, en los fibroblastos humanos modelo de enfermedad de Lafora previamente mencionados (García-Giménez et al., 2013). La desregulación de la actividad de Trx1 en estas condiciones contribuye a aumentar el estrés oxidativo, lo cual encaja perfectamente con los resultados descritos más arriba. Estos trabajos son ejemplos muy concretos de la relación existente entre el estrés oxidativo celular y la regulación de la proteostasis dentro de la célula, y cómo ambos procesos se retroalimentan de manera fisiopatológica, como la pescadilla que se muerde la cola, en el contexto de la epilepsia de Lafora, información que se puede encontrar ampliada en (Romá-Mateo, Aguado, García-Giménez, Knecht, et al., 2015).
Es difícil, no obstante, definir la relación causa-efecto entre las alteraciones moleculares recién descritas y las manifestaciones clínicas de la enfermedad; a pesar de ello, la aparición de estrés oxidativo como consecuencia de las crisis tónico-clónicas o de las mioclonías está bien documentada (Vezzani et al., 2023), y además existe otra vía de respuesta celular directamente vinculada a los síntomas neurológicos presentes tanto en enfermedad de Lafora como en otro tipo de epilepsias. Se trata de la respuesta inflamatoria, otro de los últimos frentes que se han abierto para aportar importantísima información de cara al desarrollo de biomarcadores y estrategias terapéuticas en la epilepsia de Lafora.
Las crisis epilépticas conllevan a la liberación de ATP (adenosina trifosfato) y glutamato, así como especies reactivas de oxígeno (ROS). Estas señales desencadenan la activación de la microglía y los astrocitos, y finalmente a la producción de mediadores proinflamatorios como IL-1β (interleucina 1beta), IL-6 (Interleucina 6), TNF (Factor de Necrosis Tumoral), quimiocinas o Lcn-2 (lipocalina-2), entre otros (Figura 3). La presencia de marcadores proinflamatorios en las células de la glía promueve la hiperexcitabilidad neuronal: se incrementan los niveles de glutamato en el sistema nervioso central (SNC) debido a la disminución de la actividad de los transportadores de glutamato localizados en los astrocitos, y además se produce un aumento de la liberación de este por parte de los astrocitos y la microglía (Figura 3). No solo eso, sino que también promueven la disminución de la neurotransmisión inhibitoria GABAérgica (Joensuu et al., 2014; Okuneva et al., 2015, 2016; Tegelberg et al., 2012) [revisado en (Sanz y Garcia-Gimeno, 2020; Villasana-Salazar y Vezzani, 2023)].
Por tanto, cada vez parece estar más claro que la producción de mediadores neuroinflamatorios en las células gliales favorecen las crisis epilépticas al mismo tiempo que las crisis generan un daño celular que favorece la producción de mediadores inflamatorios. En definitiva, ambos procesos, neuroinflamación e hiperexcitabilidad neuronal, forman un circuito cerrado en el que ambos procesos se retroalimentan uno sobre el otro.
Respecto a la enfermedad de Lafora, diferentes autores han demostrado la presencia de microglía y astrocitos reactivos en modelos de ratón de la enfermedad (López-González et al., 2017; Rai et al., 2017; Turnbull et al., 2011; Valles‐Ortega et al., 2011), sugiriendo la presencia de neuroinflamación en estos modelos. Reforzando la hipótesis de la neuroinflamación se ha descrito que el transportador de glutamato astrocítico (GLT-1/EAAT2) se internaliza más rápidamente al interior celular, con lo que queda menos en la superficie para ejercer su función (Muñoz-Ballester et al., 2016; Perez‐Jimenez et al., 2021), dando lugar a niveles elevados de glutamato extracelular, que favorecería la hiperexcitabilidad neuronal y las crisis epilépticas. Como se explica en este mismo párrafo, la alteración en los niveles de glutamato mediado por los astrocitos promueve las crisis y estas a su vez favorecen la neuroinflamación. En sentido contrario, la neuroinflamación produce una alteración en los niveles de glutamato. Por tanto, estos trabajos sugieren que la neuroinflamación juega un papel importante en el fenotipo de Lafora. Esta hipótesis ha sido confirmada recientemente al describirse la neuroinflamación como un sello distintivo más de la enfermedad de Lafora. En un primer estudio se realizó un análisis de RNAseq de cerebro completo en los ratones modelo de la enfermedad de Lafora (Epm2a-/- y Epm2b-/-) en el que se observó que el cerebro de estos ratones presentaba mayores niveles de expresión de algunos genes proinflamatorios, comparado con los ratones control. Estas diferencias se observaron ya a 3 meses de edad, en el caso de algunos marcadores (CXCL10) y aumentaba con la edad de los ratones (Lahuerta et al., 2020).
En un trabajo reciente, ampliando los conocimientos en el contexto de la neuroinflamación y la enfermedad de Lafora, nuestro grupo ha podido elucidar las rutas responsables de la sobreexpresión de estos genes proinflamatorios encontrados por secuenciación de ARN (RNAseq). El estudio se centró en hipocampo, por ser una región en la que existe una elevada expresión de los receptores de quimiocinas (implicadas en inflamación), además de por ser una zona altamente relacionada con epilepsia. Sorprendentemente, se encontraron niveles elevados de TNF, en ratones Epm2b-/- de 16 meses y se hipotetizó que esta molécula proinflamatoria es el principal regulador y desencadenante de la activación de una serie de rutas de señalización proinflamatorias, responsables de la neuroinflamación en la enfermedad de Lafora. Confirmando esta hipótesis, se detectaron elevados niveles de p65 y las MAPK (del inglés MAP Kinases) P38 y ERK (del inglés Extracellular signal Regulated Kinase). El aumento en p65 se correlacionó con un aumento en la ruta no canónica del inflamasoma, estando esta misma ruta alimentada por otras moléculas que se han encontrado elevadas en este modelo de ratón (P38, STAT1) (Rubio et al., 2023). La forma no canónica de inflamasoma activa la muerte celular por un proceso alternativo a la apoptosis denominado piroptosis. Así, se confirmó que en este modelo existen formas alternativas a la apoptosis y dependientes de inflamación, tales como las mediadas por caspasa-8 (necroptosis) y caspasa-11 (piroptosis). Nuestros resultados son consonantes con los obtenidos en otro modelo de ratón de epilepsia mioclónica progresiva (EPM1), en el que tras eliminar el gen responsable de esta enfermedad (Cstb-/-), se han descrito también elevados niveles de caspasa-11 como consecuencia de un aumento en p65 y se ha sugerido también la muerte celular por piroptosis como forma de muerte celular dependiente de inflamación (Maher et al., 2014). Además de TNF como regulador maestro de la inflamación en los ratones Epm2b-/-, también describimos la cascada de IL-6/JAK2 como una de las rutas responsable del estado de neuroinflamación presente en este modelo de enfermedad de Lafora (Rubio et al., 2023).
Hay que destacar que estos mediadores de inflamación, tal como se demuestra en el citado trabajo, se expresan en células de la glía (microglía y, principalmente, en astrocitos). Este resultado confirma los estudios previos realizados por secuenciación de ARN en los modelos de ratón Epm2a-/- y Epm2b-/- en donde se describe a la glía como las células responsables de la elevada expresión de genes proinflamatorios (Lahuerta et al., 2020). En consecuencia, los modelos de Lafora parecen confirmar el mecanismo encontrado en otras epilepsias en la que se ha descrito también la presencia de neuroinflamación (Sanz y Garcia-Gimeno, 2020; Villasana-Salazar y Vezzani, 2023).
Continuando con el papel que ejerce la neuroinflamación en el fenotipo de las epilepsias mioclónicas, la acción combinada de ambos reguladores proinflamatorios descritos en los ratones Epm2b-/- (TNF e IL-6) tiene un papel importante en la extravasación de células del sistema inmune de la sangre periférica al sistema nervioso central. Esto pudo confirmarse en este mismo trabajo (Rubio et al., 2023), en el que por primera vez se ha descrito la presencia de linfocitos CD3+, CD4+ y CD8+ en el hipocampo de los ratones Epm2b-/- a 16 meses de edad. Esta observación lleva a pensar que la permeabilidad de la barrera hematoencefálica en estos ratones se encuentra alterada y quizás la presencia de estos linfocitos periféricos puede exacerbar el estado neuroinflamatorio presente en el modelo. Además, del mismo modo que células del sistema inmune periférico son capaces de penetrar en el cerebro, en sentido contrario estos marcadores inflamatorios aumentados en el hipocampo podrían difundir a la sangre, sirviendo así de biomarcadores de la enfermedad. Este fenómeno se describe en detalle en el siguiente apartado.
DESARROLLO DE NUEVOS BIOMARCADORES PARA LA ENFERMEDAD DE LAFORA: UNA NUEVA GENERACIÓN DE CHIVATOS MOLECULARES
El abordaje clínico de esta compleja patología está cada vez más vinculado a la disponibilidad de herramientas diagnósticas y pronósticas que presenten altos índices de sensibilidad y especificidad, las cuales a menudo están basadas en el desarrollo y utilización de lo que se conoce como biomarcadores. Un buen biomarcador puede ser una medida de un parámetro biológico, la presencia o cuantificación de una o un conjunto de moléculas, que den idea de la diferencia entre un estado normal y patológico, y que permitan el seguimiento y monitorización de la enfermedad. Para poder cumplir adecuadamente con este objetivo, un buen biomarcador debe de ser sensible y específico, de forma que se pueda medir sus cambios de manera muy concreta y su presencia y cantidad sea un fiel reflejo de un estado patológico concreto; debe de ser relativamente sencillo y rápido de medir, de forma no invasiva y poco costosa; y su medición debe ayudar a tomar decisiones, a menudo con implicaciones pronósticas, de relevancia y utilidad para la práctica clínica. Para la enfermedad de Lafora, dada la poca disponibilidad de pacientes que permita realizar estudios clínicos de gran calado, todavía no se dispone de una gran batería de biomarcadores, pero durante los últimos años se han estado haciendo avances, gracias a los numerosos descubrimientos a nivel celular y molecular de la fisiopatología de la enfermedad, en la definición de ciertas moléculas que podrían cumplir con los criterios requeridos para funcionar como buenos biomarcadores.
Citoquinas y quimiocinas
Las citoquinas constituyen una familia de proteínas solubles que son liberadas por el sistema inmune para la activación de la respuesta innata y adaptativa como medio de defensa frente a patógenos. Numerosos análisis por métodos inmunohistoquímicos y transcriptómicos han demostrado que las citoquinas proinflamatorias, como IL1β, TNF o HMGB1 (High Mobility group Box 1), así como sus receptores, están aumentados durante las crisis epilépticas (Vezzani et al., 2019). A su vez, bajo condiciones de inflamación, se incrementa la expresión de quimiocinas, una subcategoría de citoquinas que promueven la respuesta inmunitaria en sitios específicos en los que hay inflamación. Las quimiocinas son proteínas pequeñas de entre 8 y 14KDa que a su vez activan rutas proinflamatorias como la activación de las MAPK (MAP kinases) o las rutas de señalización AP-1 (proteína activadora 1) y NF-kB (factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas)(Sanz y Garcia-Gimeno, 2020). Es por todo ello que resulta interesante estudiar estas moléculas como posibles dianas terapéuticas contra las crisis epilépticas y/o como biomarcadores de la progresión de la enfermedad.
Nuestro trabajo previo con RNAseq en muestras de cerebro de ratones modelo de enfermedad de Lafora demostró la presencia elevada de quimiocinas como CXCL10, CCL2, CCL5, CCL12 y LCN2, así como las citoquinas TNF, IL-6 e IL-1β (Lahuerta et al., 2020). Posteriormente, en un trabajo reciente, se ha confirmado que las citoquinas TNF e IL-6 están elevadas en hipocampo de ratones Epm2b-/- de 16 meses siendo ambas reguladoras maestras y activando una serie de cascadas de señalización que inician procesos de inflamación (Rubio et al., 2023). Además, pudimos observar que los niveles de estas citoquinas y quimiocinas aumentan con la edad de los ratones (Lahuerta et al., 2020). Tan solo la citoquina CXCL10 se sobreexpresa a los 3 meses de edad en los ratones modelo de Lafora, comparado con los ratones silvestres control. Por el contrario, el resto de las citoquinas y quimiocinas no se encontraron elevadas hasta los 7 meses de edad. El hecho de que la expresión de citoquinas y quimiocinas proinflamatorias aumente con la edad de los ratones nos sugiere que la neuroinflamación es progresiva y posiblemente una consecuencia de la toxicidad producida por la presencia de los cuerpos de Lafora. Así, a los tres meses de edad, cuando ya se observa la formación de los cuerpos de Lafora, se detecta sobreexpresión de CXCL10, como uno de los promotores iniciales de la inflamación. CXCL10 se expresa mayoritariamente en astrocitos (Lahuerta et al., 2020), y es uno de los mayores mediadores implicados en la reactividad astrocitaria, actuando tanto de forma paracrina como autocrina. Se ha descrito que la acción combinada de CXCL10 y su receptor CXCR3 (CXC receptor 3) aumenta los niveles de calcio intracelular, lo cual desencadena citotoxicidad. Al ser una de las primeras moléculas cuya sobreexpresión aparece a una edad temprana, estaría contribuyendo a la excitabilidad de los astrocitos y a la neuroinflamación.
Cuando las citoquinas, las quimiocinas y otros mediadores proinflamatorios se expresan a determinados niveles en el cerebro, estas moléculas pueden difundir del sistema nervioso central a la sangre periférica, siendo fáciles de detectar en una muestra de sangre y por tanto sirviendo como biomarcadores para estudiar la progresión de la enfermedad. El hecho de que se haya detectado presencia de células del sistema inmune periférico, que han infiltrado el parénquima cerebral (Rubio et al., 2023) en los ratones Epm2b-/-, sugiere que puede existir una alteración en la permeabilidad de la barrera hematoencefálica de estos ratones. Para comprobar si existe también infiltración de moléculas proinflamatorias en sentido contrario (hacia la sangre) se analizó en el suero de estos ratones las siguientes quimiocinas: LCN2, CXCL10, CCL5, C3, CCL20, S100b y HMGB1. Las 5 primeras habían sido previamente encontradas en altos niveles en los ratones modelo de enfermedad, y las dos últimas son lo que se denomina alarminas, y se han encontrado aumentadas en otras epilepsias (Miller & Blanco, 2021; Simonato et al., 2021; Terrone et al., 2018, 2019). En animales de 12 meses de edad, solo los niveles de CXCL10 y S100b se encontraron elevados en el suero de los ratones Epm2b-/-(Rubio et al., 2023). Estos resultados apuntan a que CXCL10 y S100b pueden ser consideradas como biomarcadores de la progresión de la enfermedad, por lo que es de indudable interés analizar estas y otras biomoléculas en suero de pacientes, con el fin de corroborar las observaciones descritas en los modelos de ratón de enfermedad de Lafora.
microARN
Los microARN son pequeñas moléculas de ARN no codificante, de entre 18 y 25 nucléotidos de longitud, que participan de la regulación de la expresión génica. Tras un complejo proceso de síntesis y maduración, los microARN son capaces de unirse a moléculas de ARN mensajero, en base a la complementariedad entre secuencias nucleotídicas; esta unión deriva habitualmente en la desestabilización del mensajero o el impedimento de acceso de la maquinaria traduccional, produciendo por tanto una inhibición de la expresión génica a nivel post transcripcional. La participación de los microARN en la regulación constitutiva de la expresión génica en toda clase de tipos celulares y participando de manera crítica en la regulación de multitud de procesos está ampliamente documentada, así como la relevancia de alteraciones en este nivel de regulación para el desarrollo de numerosas patologías, por lo que los microARN se han perfilado como un importante frente de investigación en biomedicina, constituyendo al mismo tiempo potenciales dianas terapéuticas. No obstante, estas moléculas proporcionan un punto adicional de interés por el hecho de que son secretados al exterior celular, bien por medio de exosomas, en forma libre o unidos a proteínas, para su transporte a través del plasma, llegando a ser capaces de ser incorporados por tejidos y órganos distantes del lugar en que fueron secretados y regulando la expresión génica en dichos destinos. La detección y cuantificación de microARN en muestras de plasma es un método de relativa sencillez y bajo coste, muy poco invasivo, lo cual ha propiciado que se haya desarrollado numerosos estudios y propuestas para utilizar los patrones de microARN circulantes como biomarcadores de enfermedad, encontrándose perfiles específicos —lo que se conoce como firmas de microARN— correspondientes a estados de enfermedad o incluso a diferentes fases de la misma patología (Ho et al., 2022; Simonson y Das, 2015).
Existe evidencia de una estrecha relación entre los mecanismos moleculares alterados en procesos que cursan con epilepsia y una desregulación de la expresión de microARN, por lo que algunos tipos específicos han sido propuestos como potenciales biomarcadores para ciertos tipos de epilepsia (Bauer et al., 2020; Ünalp et al., 2022; Wang & Zhao, 2021; Whitlock et al., 2022). En el caso de enfermedad de Lafora, la información al respecto de las alteraciones a nivel de microARN son muy escasas. Recientemente nuestro grupo ha descubierto, en cerebros de ratones modelo de enfermedad, un incremento muy significativo de dos especies concretas de microARN: miR-146a y miR-155 (Romá-Mateo et al., 2023). Este incremento es progresivo según avanza la edad de los ratones, y correlaciona con alteraciones importantes en genes implicados en la regulación de la respuesta a estrés oxidativo y la inflamación, corroborando los datos recabados a nivel de estudios transcriptómicos y bioinformáticos en dichos modelos (Jabato et al., 2021; Lahuerta et al., 2020). Estos microARN han sido previamente estudiados en el contexto de otras epilepsias (An et al., 2016; Fu et al., 2019; Huang et al., 2022; Liu et al., 2022), e incluso se han propuesto como posibles dianas terapéuticas, como veremos en el apartado siguiente.
NUEVAS APROXIMACIONES TERAPÉUTICAS: EL HORIZONTE DE LA INVESTIGACIÓN EN ENFERMEDAD DE LAFORA
Fármacos generales anticrisis
Como ya se ha indicado anteriormente, todavía no existe un tratamiento eficaz para la enfermedad de Lafora. Los pacientes se tratan inicialmente con fármacos anticrisis con el fin de aliviar estos síntomas. Este tratamiento consiste en la administración de combinaciones tales como ácido valproico, benzodiacepinas, piracetam, zonisamida y levetiracetam, evitando el uso de carbamazepina, gabapentina, vigabatrina, lamotrigina, tiagabina y fenitoina, ya que estos últimos pueden exacerban las mioclonías o la ataxia (Michelucci et al., 2016; Monaghan y Delanty, 2010; Turnbull et al., 2016; Vorderwülbecke et al., 2022). Desgraciadamente, estos fármacos funcionan inicialmente, pero pronto los pacientes se vuelven resistentes a su acción. Se han descrito efectos beneficiosos de nuevos fármacos anticrisis tales como perampanel, un antagonista de los receptores postsinápticos de glutamato tipo AMPA (Dirani et al., 2014; Obara et al., 2021; Schorlemmer et al., 2013). Sin embargo, en ciertos casos se describieron efectos adversos del producto (e.g., irritabilidad), que aconsejaron la suspensión del tratamiento (Goldsmith y Minassian, 2016).
Dieta cetogénica
La dieta cetogénica se ha probado en diferentes tipos de epilepsias con efectos beneficiosos. Sin embargo, el seguimiento de este tipo de dieta no es muy aceptado por muchos pacientes por sus malas características organolépticas, además de efectos secundarios como pérdida de peso, estreñimiento, y elevación de los niveles de VDL y colesterol en sangre (Verhoog et al., 2020). Dado que este tipo de dieta suele utilizarse en caso de epilepsias farmacorresistentes y/o catastróficas, se ha intentado utilizar en enfermedad de Lafora, pero no se logró detener la progresión de la enfermedad (Cardinali et al., 2006).
Tratamiento para evitar las mutaciones sin sentido
En la enfermedad de Lafora se han definido mutaciones tanto en el gen EPM2A como EPM2B, que conducen a una parada prematura de la traducción, originando formas truncadas de las proteínas que no son funcionales; basándose en esta característica, se han utilizado fármacos, como la gentamicina, que promueven un salto en el codón de parada prematuro, pero con un éxito muy limitado (Delgado-Escueta y Bourgeois, 2008).
Fármacos de reposicionamiento
Los fármacos de reposicionamiento son aquellos que se están utilizando para aliviar o curar una enfermedad concreta, pero que pueden tener efectos beneficiosos en otras patologías diferentes. La enfermedad de Lafora se ha beneficiado de este tipo de estrategia y en ensayos preclínicos en ratón, ciertos compuestos han demostrado su efecto beneficioso. Así, compuestos como el 4-fenil-butirato (de uso en trastornos del ciclo de la urea) (Berthier et al., 2016), la trehalosa (de uso como agente humectante) (Berthier et al., 2016; Sinha et al., 2021b), el selenato sódico (de uso en enfermedades con deterioro cognitivo y demencias) (Sánchez-Elexpuru et al., 2017), la dexametasona (de uso como corticoide anti-inflamatorio general) (Sinha et al., 2021a), el propranolol (de uso como antihipertensivo y mejora de la frecuencia cardíaca) (Mollá et al., 2021), el canabidiol (de uso para aliviar el dolor muscular) (Aso et al., 2020), la memantina (de uso en la enfermedad de Alzheimer) (Mollá et al., 2021), y la minociclina (de uso como antibiótico en el tratamiento de enfermedades respiratorias) (Mollá et al., 2021), han mostrado un efecto beneficioso en modelos murinos de enfermedad de Lafora. Sin embargo, su uso en pacientes no ha sido descrito hasta el momento, por lo que ninguno de los anteriores fármacos ha podido demostrar su eficacia en el control de la enfermedad.
Una mención especial en este apartado merece la metformina (de uso habitual como antidiabético oral). Este compuesto demostró su efecto beneficioso en modelos de ratón (Berthier et al., 2016) y también se ha utilizado en el tratamiento de pacientes. En un primer estudio, la metformina enlenteció la progresión de la enfermedad en tres pacientes de enfermedad de Lafora de un total de doce; los autores sugirieron que la falta de respuesta de los nueve restantes podría ser debido a su estado avanzado de la enfermedad (Bisulli et al., 2019). En un estudio posterior, con ocho pacientes en estadios iniciales de la enfermedad, la administración de metformina mostró un claro efecto beneficioso al lograr un retraso notable en la evolución de la enfermedad. Los pacientes tratados mostraron una gran mejoría en su capacidad de habla, higiene y aseo personal (Burgos et al., 2023). Estos trabajos resaltan la importancia de la administración temprana de metformina para conseguir un enlentecimiento de la evolución de la enfermedad, y la definen como un medicamento de elección para el tratamiento inicial de la enfermedad de Lafora, hasta que se validen tratamientos futuros más eficaces.
Estrategias para disminuir la síntesis de glucógeno
Como se ha indicado más arriba, el hecho significativo de la enfermedad de Lafora es la acumulación de formas aberrantes de glucógeno insolubles (poliglucosanos) en el cerebro de los pacientes. La hipótesis actual más aceptada es que estos acúmulos son los responsables de la fisiopatología de la enfermedad (Markussen et al., 2021). Por ello, diferentes grupos han propuesto diferentes estrategias con el fin de prevenir su aparición. La primera estrategia fundamentada en este objetivo consistió en diseñar compuestos químicos que funcionaran como inhibidores de la enzima responsable de la síntesis inicial de los poliglucosanos —la ya indicada glucógeno sintasa— que se encuentra en el cerebro (en concreto, la isoforma GYS1). Como resultado de los primeros rastreos, se identificó un conjunto de moléculas con una alta capacidad para inhibir la isoforma cerebral GYS1 (Tang et al., 2020). En estos momentos se está a la espera de la validación del uso de estos compuestos en modelos animales.
La segunda estrategia ha consistido en prevenir la expresión del gen GYS1 en el cerebro mediante el desarrollo de oligonucleótidos antisentido (AntiSense Oligonucletides, ASO). Para ello, se han diseñado unos GYS1-ASO que se administraron por vía intracerebroventricular en ratones, demostrando un efecto reductor sobre la actividad de GYS1 y el acúmulo de glucógeno. De hecho, si la administración se realizaba en edades tempranas (un mes), se prevenía la formación de poliglucosanos en el cerebro de los ratones; sin embargo, si se administraban en edades tardías no se producía una disminución de los poliglucosanos ya existentes (Ahonen et al., 2021). Una estrategia alternativa consistió en la disminución de la expresión del gen GYS1 por introducción de un microARN específico usando virus adenoasociados (AAV9-miRNA-GYS1) por vía intracerebroventricular, obteniéndose una disminución en la expresión del gen GYS1 y en la cantidad de poliglucosanos (Gumusgoz et al., 2022). Una tercera variante de esta estrategia consistió en introducir disrupciones en el gen GYS1 mediante el uso de la tecnología CRISPR-Cas9. Este procedimiento fue capaz de disminuir la expresión de GYS1 y la cantidad de poliglucosanos presentes (Gumusgoz et al., 2021) aunque, por el momento, todavía no se ha utilizado ninguna de las tres versiones de esta estrategia en pacientes.
Estrategias para degradar los poliglucosanos
Una estrategia alternativa para disminuir los niveles de poliglucosanos consiste en la expresión de enzimas que se encarguen de degradarlos. Los poliglucosanos son polímeros de glucosa encadenados y estrechamente compactados: la utilización de enzimas como la alfa-amilasa o la alfa-glucosidasa podrían romper estas cadenas y hacerlas susceptibles de ser degradadas. Con el fin de hacer llegar cualquiera de estas dos enzimas al cerebro, se ha diseñado una proteína quimera donde la enzima responsable de la degradación de los poliglucosanos se fusiona a un fragmento de inmunoglubulina G y un péptido penetrante, para así poder acceder al interior de las células del parénquima cerebral (alfa-amilasa, VAL0417; alfa-glucosidasa, VAL1221). La vía de administración sigue siendo intracerebroventricular con el fin de salvar la barrera hematoencefálica. La administración de estos compuestos en ratones modelo de la enfermedad redujo los niveles de poliglucosanos (Austin et al., 2019; Brewer et al., 2019), pero una vez más, todavía no se ha informado de su aplicación en pacientes humanos.
Terapia génica
Dado que la enfermedad de Lafora se hereda con carácter recesivo, en los últimos años se ha empezado a diseñar estrategias con el fin de introducir un gen normal en las células afectadas. Una primera aproximación ha sido la del uso de liposomas catiónicos conjugados con plásmidos codificantes para laforina. Sin embargo, usando células en cultivo se observó que la eficiencia de introducción era bastante baja (Vemana et al., 2021). Recientemente se ha propuesto el uso de virus adenoasociados AAV9 conteniendo el gen de laforina o malina, y con el fin de mejorar la eficiencia de transducción, también se ha propuesto la utilización de ultrasonidos focalizados de alta intensidad, cuyo uso ha mejorado la distribución del AAV9 en el cerebro en otras patologías. Sin embargo, todavía no hay referencias en la literatura sobre la aplicación de estas técnicas en la enfermedad de Lafora (Mitra et al., 2022).
Agonistas y antagonistas de microARN
Como se ha explicado en el apartado anterior, la participación de los microARN en la regulación de mecanismos celulares cuya alteración subyace a multitud de procesos patológicos los ha convertido a su vez en potenciales dianas terapéuticas. Por la propia naturaleza molecular de los microARN, es relativamente sencillo diseñar oligonucléotidos sintéticos que bien mimeticen la secuencia de un microARN (agonistas) o constituyan una secuencia complementaria del mismo (antagonistas): en el primer caso, la adición de dicho oligonucleótido produce un efecto de sobreexpresión, mientras que en el segundo se produciría una inactivación de los efectos mediados por los microARN al producirse la unión del antagonista y su diana, impidiendo ejercer la función fisiológica normal. Esta estrategia de diseño relativamente simple, que ha demostrado funcionar bien a nivel experimental en líneas celulares, adquiere complejidad cuando se intenta aplicar a nivel de organismo completo, por lo que el desarrollo de terapias basadas en el uso de agonistas y antagonistas de microARN (también denominados agomirnas y antagomirnas) ha sido muy limitado hasta la fecha actual. Especialmente difícil es la aplicación de este tipo de terapias para enfermedades que cursan con deterioro del sistema nervioso central, con la dificultad añadida de la necesidad de cruzar la barrera hematoencefálica para ejercer un efecto clínico significativo sobre los procesos neuroinflamatorios para los que se ha descrito un importante papel para numerosos microARN. No existen terapias basadas en agomirnas o antagomirnas para el tratamiento de enfermedad de Lafora, pero es interesante destacar que algunos trabajos de silenciamiento de miR-155 en modelos experimentales han mostrado un efecto beneficioso (Fu et al., 2019), e incluso se ha conseguido reducir el número de crisis y la severidad de las crisis en modelos de ratón de epilepsia tras la inoculación intranasal de un antagonista para miR-155 (Zhou et al., 2020). Sin duda el estudio de los métodos de silenciamiento de microARN como posibles terapias para frenar el avance de la epilepsia se perfila como un interesante frente de investigación a nivel preclínico.
En resumen, diferentes aproximaciones terapéuticas se han ensayado en ratones modelos de enfermedad de Lafora, demostrando su efecto beneficioso (Tabla 1). Sin embargo, el paso a su utilización en la clínica todavía no se ha producido. Esperemos que en un futuro temprano se puedan implementar alguna de estas estrategias en pacientes con el fin de conocer su potencialidad como posible tratamiento. Mientras tanto, hasta el momento, solo la metformina ha demostrado su efecto enlentecedor en la evolución de la enfermedad en pacientes. En nuestra opinión, un tratamiento combinado de metformina y alguna de las aproximaciones descritas en esta sección, podría ser la opción terapéutica a seguir para minimizar los efectos deletéreos de esta terrible enfermedad.
Tabla 1: Aproximaciones terapéuticas en la enfermedad de Lafora.
| Aproximación terapéutica | Producto | Utilización en Lafora |
| Fármacos anticrisis | Ácido valproico, benzodiacepinas, piracetam, zonisamida, levetiracetam, perampanel | Humanos |
| Dieta cetogénica | Dieta cetogénica | Humanos |
| Salto de codón parada prematura | Gentamicina | Humanos |
| Fármacos de reposicionamiento | 4-fenil-butirato, trehalosa, selenato sódico, dexametasona, propranolol, canabidiol, memantina, minociclina | Modelos ratón Lafora |
| Metformina | Modelos ratón Lafora y Humanos | |
| Disminución síntesis glucógeno | Inhibidores GYS1,
Oligonucleotidos antisentido GYS1, microARN GYS1, CRISPR-Cas9-GYS1 |
Modelos ratón Lafora |
| Degradación poliglucosanos | VAL0417 (alfa-amilasa)
VAL1221 (alfa-glucosidasa) |
Modelos ratón Lafora |
| Terapia Génica | Liposomas catiónicos | Líneas celulares
|
LA ENFERMEDAD DE LAFORA, UN PROBLEMA DE SALUD DE INTERÉS PARA LA SOCIEDAD
La enfermedad de Lafora es el ejemplo más paradigmático de una enfermedad ultrarara, al afectar entre 1-9 personas por millón de individuos (según Orphanet). Al igual que otras enfermedades raras, sufre de los tres grandes hándicaps que caracterizan a estas dolencias: i) pobre conocimiento científico de su fisiopatología; ii) falta de acceso a un rápido diagnóstico; iii) ausencia de un tratamiento que cure o alivie la enfermedad. A pesar del tremendo esfuerzo que se ha hecho en los últimos años en comprenderla, las bases moleculares de las alteraciones fisiopatológicas que acompañan a la enfermedad de Lafora todavía se desconocen. En cuanto a su diagnóstico, se ha avanzado gracias a la accesibilidad de las pruebas genéticas para secuenciar los genes diana. No obstante, en algunos casos no se detecta la mutación responsable con esta tecnología, debido a la presencia de inserciones y/o deleciones grandes en el genoma del paciente. Respecto del tratamiento, como se ha expuesto previamente, a día de hoy existen una serie de aproximaciones terapéuticas diseñadas para aliviar la patología de la enfermedad, y queda claro que el efecto de estas estrategias es mayor si la enfermedad está en sus inicios. Por ello es importante realizar pruebas genéticas a familiares de afectos (hermanos) con el fin de diagnosticar la presencia incipiente de la enfermedad, para empezar el tratamiento lo antes posible.
La enfermedad de Lafora es un desorden neurológico muy grave. Desde este foro, queremos dar las gracias a las asociaciones de pacientes, pues saben de primera mano lo que supone tener un paciente de Lafora en su familia y, por ello, pueden trasladar su conocimiento, empatía y consuelo a otras familias en situaciones similares. Nos gustaría romper una lanza a favor de la Asociación Española para Vencer la Enfermedad de Lafora (AEVEL; https://www.aevel.org/) y a la Asociación Americana de la Enfermedad de Lafora (Chelsea’s Hope; https://chelseashope.org/), que hacen un trabajo ímprobo en la ayuda a las familias de pacientes con la enfermedad de Lafora. Estas asociaciones son las que tenemos más cerca, pero queremos extender nuestro agradecimiento al resto de asociaciones de familiares de pacientes con enfermedad de Lafora del mundo, por su desinteresada labor en favor de las familias con pacientes con la enfermedad de Lafora. No podemos dejar de resaltar que todas estas personas constituyan parte de nuestra sociedad, y el hecho de que constituyen una fracción muy pequeña de la población no es razón para olvidar su problemática o mermar los esfuerzos para encontrar una cura o, al menos, intentar mejorar sus condiciones de vida.
REFLEXIONES FINALES
En este trabajo hemos pretendido hacer una revisión de la complejidad molecular asociada a la enfermedad de Lafora y las estrategias terapéuticas que están surgiendo en los últimos años. Es indudable que desde que en 1911 se describiera esta enfermedad por Gonzalo Rodríguez Lafora, se ha avanzado enormemente; las dos últimas décadas han sido especialmente prolíficas en cuanto a la cantidad de trabajos y evidencias experimentales que se han hecho eco de todas las alteraciones características de la enfermedad, sirviendo de base para numerosos procedimientos y ensayos con un potencial enorme para mejorar la calidad de vida de los pacientes. Nos gustaría pensar que alguna de las estrategias terapéuticas actuales o las que se puedan plantear en un futuro, darán su fruto y eventualmente conseguirá ver la luz un tratamiento para esta enfermedad tan terrible que alivie la vida de los pacientes y la de sus familias.
Declaración de ausencia de conflictos de intereses
Los autores declaran que no tienen ningún conflicto de interés.
AGRADECIMIENTOS
Los autores quisieran agradecer las siguientes fuentes de financiación: Ministerio de Ciencia e Innovación (PID2020-112972RB-I00 y PID2020-119127RA-I00), Fundació La Marató TV3 (202032), y National Institutes of Health (P01NS097197).
Este trabajo se lo dedicamos a dos pacientes de Lafora que conocemos directamente: a Laura, que nos deleita con sus canciones, y a Fátima que nos conquistó con su sonrisa.


