El sistema nervioso, el centro de mando de nuestro cuerpo, es un sistema que comienza a formarse desde etapas muy tempranas del desarrollo embrionario y continúa madurando durante los primeros años de vida. Este proceso, tan fascinante como delicado, depende de una compleja interacción entre factores ambientales y biológicos, entre los que se encuentran los factores genéticos. Por tanto, la genética puede jugar un papel fundamental en el desarrollo de las enfermedades neurológicas pediátricas, un grupo de trastornos que afectan al cerebro, la médula espinal, los nervios y otros componentes del sistema nervioso en pacientes pediátricos.

A continuación, veremos algunas de las principales enfermedades neuropediátricas y cómo la genética puede ayudar no solo a entender mejor su origen, sino también a mejorar el diagnóstico, personalizar los tratamientos y anticiparnos en la intervención clínica.
Epilepsias de origen genético en la infancia
La epilepsia es una condición neurológica que afecta a entre 4 y 11 de cada 1.000 infantes menores de 14 años y que está caracterizada por crisis recurrentes debido a una actividad eléctrica anormal en el cerebro. En la infancia, muchas epilepsias tienen un origen genético, especialmente aquellas que se manifiestan de forma temprana y con crisis difíciles de controlar.
Síndrome de Dravet y SCN1A
Algunas formas, como la epilepsia mioclónica severa de la infancia (síndrome de Dravet), están causadas por mutaciones en el gen SCN1A. Este gen codifica la subunidad alfa de un canal de sodio dependiente de voltaje, esencial para la correcta generación y propagación del potencial de acción en las neuronas, especialmente en las interneuronas inhibitorias. La disfunción de este canal propicia la aparición de crisis epilépticas refractarias y otros trastornos neurológicos.
Otros genes implicados: KCNQ2, STXBP1 y CDKL5
Además de SCN1A, se han identificado numerosos genes implicados en epilepsias infantiles. Entre ellos, KCNQ2, que codifica un canal de potasio involucrado en el control de la excitabilidad neuronal, se asocia tanto a epilepsias neonatales benignas como a formas graves con retraso del desarrollo.
Otro ejemplo es el gen STXBP1, clave para la liberación de neurotransmisores en la sinapsis. Las mutaciones en este gen están relacionadas con encefalopatías epilépticas tempranas caracterizadas por crisis de inicio neonatal o en los primeros meses de vida, junto con discapacidad intelectual y trastornos del movimiento.
Por su parte, el gen CDKL5 se asocia a una encefalopatía epiléptica de inicio muy precoz, con crisis de difícil control, alteraciones motoras severas y afectación cognitiva profunda.
Diagnóstico y tratamiento personalizado
En el contexto de las epilepsias infantiles, el uso de pruebas genéticas se ha convertido en una herramienta clave que va más allá de la mera confirmación diagnóstica. La identificación de la alteración genética responsable permite definir con mayor precisión el tipo de epilepsia del paciente. Esto permite reducir la incertidumbre clínica, evitar pruebas innecesarias y acortar el tiempo de diagnóstico.
En cuanto a tratamiento, conocer la base genética de un tipo de epilepsia puede ayudar a seleccionar mejor el fármaco a suministrar y reducir problemas asociados. Por ejemplo, en epilepsias asociadas a mutaciones en SCN1A está contraindicado utilizar bloqueadores de canales de sodio, ya que pueden empeorar las crisis epilépticas.
Atrofia muscular espinal (AME): genética y tratamiento
La atrofia muscular espinal (AME) es una de las enfermedades neuromusculares pediátricas hereditarias más conocidas. Se trata de una de las enfermedades neuromusculares más comunes y está presente en 2 de cada 100.000 personas. La AME afecta a las neuronas motoras de la médula espinal, responsables del control del movimiento, y causa una debilidad muscular progresiva que puede comprometer funciones vitales como la deglución o la respiración.
El gen SMN1 y la proteína SMN
La atrofia muscular espinal está causada principalmente por mutaciones en el gen SMN1, que codifica la proteína SMN (Survival Motor Neuron Protein), una proteína necesaria para la supervivencia de las neuronas motoras. Las alteraciones en SMN1 hacen que la proteína SMN se vea truncada, lo que provoca la degeneración de las neuronas motoras y la aparición de la sintomatología típica de la enfermedad.
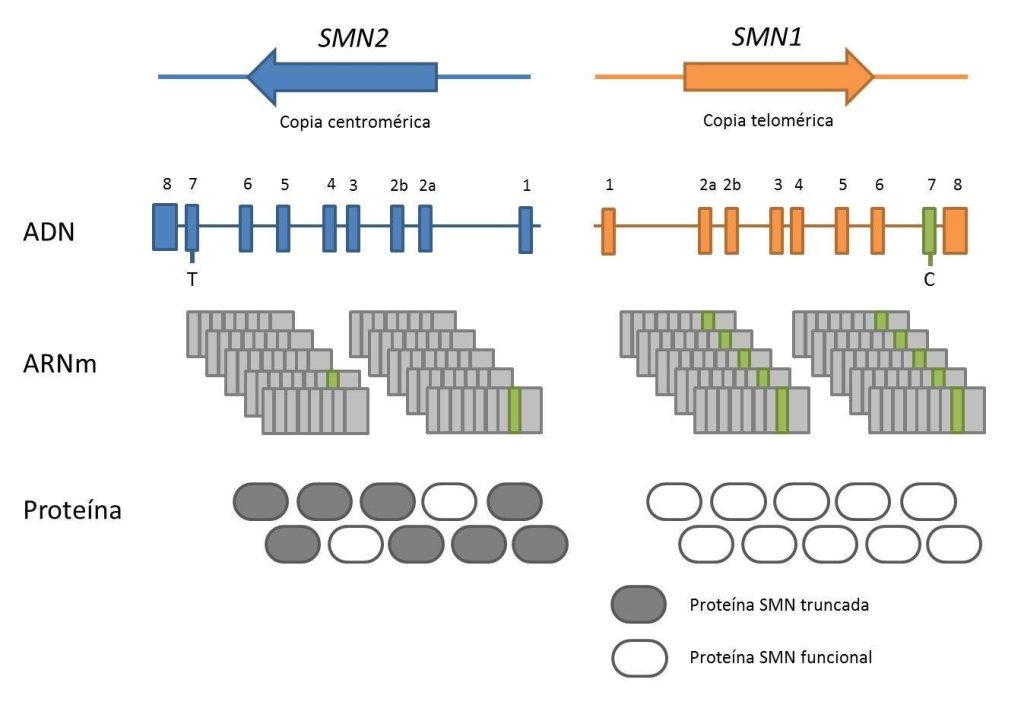
Avances en terapia génica: Zolgensma y Risdiplam
El desarrollo de terapias dirigidas ha supuesto un cambio radical en el abordaje de la atrofia muscular espinal. Uno de los hitos más relevantes ha sido la aprobación en 2019 de Zolgensma en 2019, la primera terapia génica para esta enfermedad. El tratamiento, que introduce una copia funcional del gen SMN1 en las células del paciente, ha demostrado mejoras significativas en la función motora de los pacientes.
Paralelamente, se han desarrollado tratamientos farmacológicos no invasivos como Risdiplam, un fármaco de administración oral que actúa modulando el splicing del gen SMN2, aumentando la producción de proteína SMN funcional.

Trastornos del desarrollo intelectual de causa genética
Dentro de las enfermedades neuropediátricas también se encuentran los trastornos del desarrollo intelectual, condiciones neurológicas caracterizadas por un funcionamiento intelectual significativamente inferior al promedio. Este tipo de trastornos, que suelen manifestarse en etapas tempranas de la infancia, suelen estar acompañados de limitaciones en habilidades adaptativas como la comunicación, el cuidado personal, la vida social o la autonomía del paciente.
Se estima que aproximadamente un 1% de la población española presenta algún tipo de trastorno del desarrollo intelectual. Según la Asociación Española de Pediatría, los infantes con este tipo de trastornos lo presentan en coexistencia con trastornos motores (7%), alteraciones neurosensoriales (7%), epilepsia (10%) o trastorno del espectro autista (2%)
Genética del desarrollo intelectual: Síndrome X-Frágil, síndrome de Rett y síndrome de Angelman
A nivel genético, se han identificado cientos de genes asociados a la discapacidad intelectual. Entre los ejemplos más conocidos se encuentra FMR1, cuya alteración causa el síndrome X frágil, la causa hereditaria más frecuente de discapacidad intelectual, caracterizada por alteraciones en la regulación de la traducción de proteínas sinápticas.
Otro gen clave es MECP2, asociado al síndrome de Rett, un trastorno del neurodesarrollo que afecta principalmente a niñas y se manifiesta con una regresión neurológica tras un desarrollo inicial aparentemente normal.
De igual modo, las mutaciones en el gen UBE3A se han asociado al desarrollo del síndrome de Angelman, un tipo de discapacidad intelectual severa, caracterizado por ausencia o escasez del lenguaje y alteraciones motoras.
Estos genes, entre muchos otros, cumplen funciones esenciales para el desarrollo y el mantenimiento del sistema nervioso, incluyendo la sinaptogénesis y la plasticidad neuronal, entre otros.
Los últimos avances en genética molecular están ayudando a diagnosticar y clasificar mejor los diferentes trastornos que provocan déficit intelectual. Esto ayuda a predecir la evolución clínica de cada paciente y, en ciertos casos, a orientar tratamientos personalizados.
Trastornos del espectro autista (TEA) y genética
Uno de los grupos más estudiados dentro de las enfermedades neuropediátricas son los trastornos del espectro autista (TEA), que afectan la comunicación, la interacción social y el comportamiento de aproximadamente 1 de cada 100 infantes.
Más de 100 genes asociados al TEA
Aunque no existe una única causa genética responsable de los trastornos del espectro autista, la evidencia científica indica que se trata de patologías con una base genética compleja y multifactorial. Hasta la fecha, se han identificado más de un centenar de genes asociados a un mayor riesgo de desarrollar TEA.
Entre los más conocidos están los genes SHANK3, MECP2 o SCN2A, implicados en la sinapsis neuronal y en la regulación de la expresión génica en el cerebro. No obstante, se trata de un grupo de trastornos bastante complejo en el que recientes estudios continúan aportando datos clave para entender los mecanismos genéticos detrás del desarrollo de TEA.
¿Cuándo solicitar pruebas genéticas?
En cuanto a las pruebas genéticas, no suelen realizarse en casos de TEA. Al menos, no para el diagnóstico de este trastorno, ya que suele ser fundamentalmente clínico. No obstante, los estudios genéticos pueden desempeñar un papel complementario muy relevante en determinados casos, especialmente cuando el TEA se acompaña de otros signos neurológicos o del desarrollo. En este contexto, las pruebas genéticas permiten identificar o descartar causas genéticas subyacentes, como el síndrome de Rett o el síndrome X frágil.
¿Por qué el diagnóstico genético es importante en enfermedades neuropediátricas?
Como comentábamos, los estudios genéticos se han convertido en una herramienta fundamental para comprender las bases de muchas enfermedades neuropediátricas. Y es que, gracias a las técnicas actuales como la secuenciación genómica, es posible analizar de forma simultánea un gran número de genes implicados en el desarrollo y funcionamiento del sistema nervioso
El análisis de variantes patogénicas permite identificar alteraciones genéticas concretas responsables de trastornos neurológicos de inicio temprano, incluso en casos con fenotipos complejos o solapados. Este enfoque no solo facilita un diagnóstico más preciso y temprano, sino que también contribuye a comprender mejor los mecanismos biológicos subyacentes a cada patología.
Desde el punto de vista clínico, la información genética obtenida puede ser clave para adaptar el manejo y el seguimiento del paciente, ayudando a predecir la evolución de la enfermedad, identificar posibles comorbilidades y, en determinados casos, orientar hacia tratamientos más personalizados o terapias dirigidas.
Especialízate en Genética y Neuropediatría con Genotipia
En el contexto actual de la medicina de precisión y del rápido avance de la genética clínica, es esencial que los profesionales estén formados en genética y enfermedades neurológicas pediátricas.
Por ello, desde Genotipia hemos creado un programa específico, Experto en Genética y Neuropediatría. Este programa, que profundiza en las bases genéticas de las enfermedades neurológicas y pediátricas, busca servir como guía a los profesionales que se dedican al diagnóstico y el manejo de las enfermedades neuropediátricas.
Si estás interesado en nuestra formación y necesitas más información, puedes agendar una llamada con nuestro equipo.

