
Hoy es 28 de junio, y esta es una fecha importante. Es un día para concienciar al público sobre una enfermedad que afecta a miles de personas: el síndrome de Folling, más conocido como fenilcetonuria. En efecto, el 28 de junio es el Día Mundial de la Fenilcetonuria.
¿Qué es la fenilcetonuria?
La fenilcetonuria (o PKU para abreviar) es una enfermedad heredable y rara que consiste en un aumento de la concentración de fenilalanina (Phe) en la sangre. La Phe es un aminoácido, es decir, es uno de los 22 bloques básicos con los que se construyen las proteínas.
En una persona sana la fenilalanina libre es transformada en otro aminoácido, tirosina, por una enzima llamada Fenilalanina Hidroxilasa (o PAH para abreviar). Sin embargo cuando esta enzima no se fabrica en el cuerpo o no funciona bien la Phe se acumula en la sangre y se transforma en otras moléculas parecidas y tóxicas: fenilpiruvato, fenilactato y fenilacetato. Estas moléculas tóxicas pueden alterar el desarrollo normal del cerebro, pudiendo llegar a causar problemas neuronales o psicológicos, especialmente en niños cuyo sistema nervioso aún se está desarrollando. Los pacientes con PKU también suelen ser más pálidos y en algunos casos tienen un olor parecido al de un ratón, debido a la acumulación de Phe.
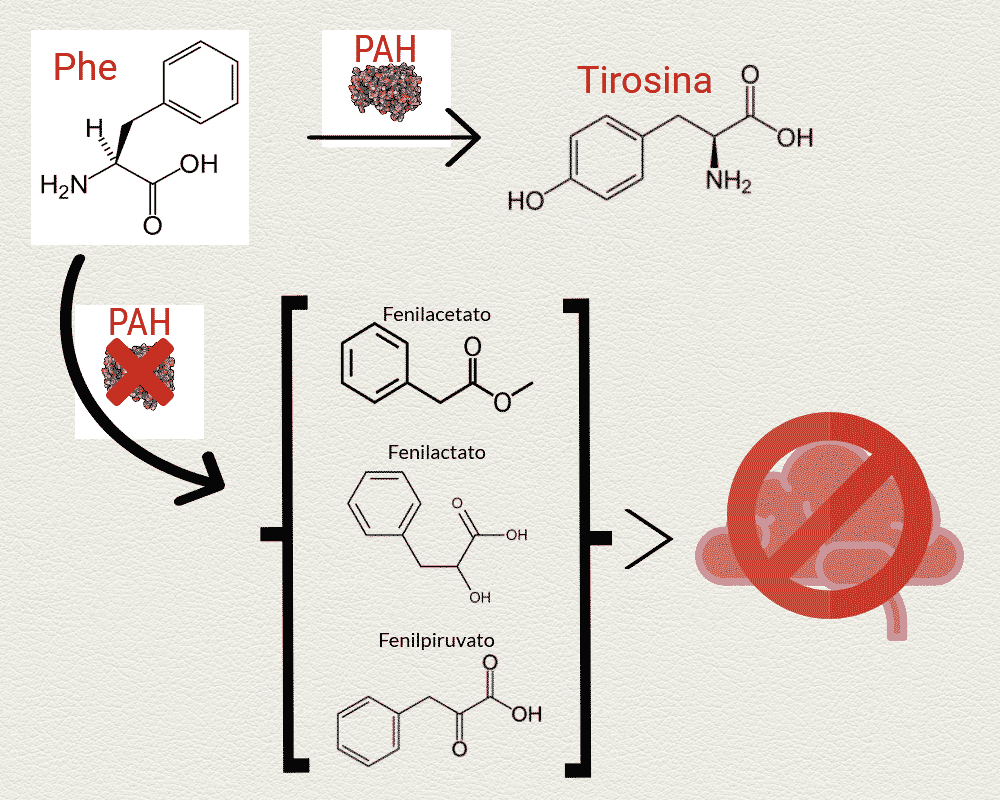
La forma más severa de fenilcetonuria se llama “PKU clásica”, y ocurre cuando la PAH está muy reducida o totalmente ausente. En este caso los niños parecen normales hasta que alcanzan los varios meses de edad. Si no reciben tratamiento, estos niños pueden desarrollar discapacidad intelectual permanente, y también son comunes las convulsiones, los problemas de comportamiento y los desórdenes psiquiátricos. Sin embargo, también existen variantes de fenilcetonuria menos severas cuando sí que existe cierta actividad de PAH.
¿Cómo se produce?
La deficiencia en la actividad de la enzima PAH que ocurre en pacientes con fenilcetonuria es debida a defectos en el gen PAH que codifica la enzima y que se encuentra en el cromosoma 12. Otros genes que intervienen en la actividad de PAH (por ejemplo, regulando su fabricación) también pueden modular la severidad de la fenilcetonuria.
Los defectos en el gen PAH que causan PKU suelen ser autosómicos recesivos. Es decir, una persona necesitaría que ambas copias del gen estuvieran defectuosas para desarrollar la enfermedad.
Qué hacer cuando tienes fenilcetonuria
Afortunadamente, la fenilcetonuria es una enfermedad muy fácil de detectar: tan solo hay que realizar un análisis de sangre y ver si hay exceso de fenilalanina. Esta es una prueba rutinaria que se realiza a casi todos los recién nacidos (la conocida prueba del talón), así que no hay que preocuparse de que la enfermedad pase desapercibida. Además, un análisis enzimático o una prueba genética pueden determinar si los padres son portadores de un gen PAH defectuoso.
Mucho menos afortunadamente, la PKU no tiene cura. Sin embargo, se pueden evitar los síntomas si se consume una dieta baja en fenilalanina. Básicamente se tiene que evitar comer alimentos con muchas proteínas (leche, huevos, carne, etc.) y alimentos que contengan el edulcorante artificial aspartamo (ácido aspártico junto con fenilalanina), que se encuentra en muchas bebidas gaseosas como la Coca-Cola o la Pepsi, en chicles, cereales o incluso en productos farmacéuticos. Por suerte, existen leches en polvo especiales, diseñadas como fuente de proteínas específicamente para personas con fenilcetonuria.
También existen medicamentos que reducen los niveles de Phe en sangre, como Palynziq o BH4, pero ambos tienen efectos secundarios y pueden producir reacciones de hipersensibilidad. Y actualmente se está desarrollando también una medicina biótica sintética, SYNB1618, que son células de E. coli modificadas genéticamente para que sean capaces de metabolizar Phe y convertirlo en compuestos inofensivos.
¿Necesitas ayuda?
Hay varias asociaciones que ayudan a gente con fenilcetonuria, como la Asociación de Fenilcetonúricos y OTM de Madrid (ASFEMA) o la Associació Catalana de PKU i altres Trastorns Metabòlics. Estas organizaciones proporcionan información sobre la enfermedad, noticias sobre su investigación o incluso recetas que se ajusten a la dieta de los pacientes con fenilcetonuria.
Un proyecto de la Universidad de Granada, además, fue la creación de www.pku.es, una página web sin anuncios con recursos gratuitos para educar a los niños sobre la fenilcetonuria. Además de contener información y ayuda como en las asociaciones mencionadas anteriormente, también han desarrollado videojuegos educativos. Es, en resumen, una página web dedicada a educar a niños y adultos sobre fenilcetonuria.
Más información
- Sobre la fenilcetonuria (en inglés): https://ghr.nlm.nih.gov/condition/phenylketonuria
- Información para pacientes: https://medlineplus.gov/spanish/ency/article/001166.htm
- Página web educativa sobre la PKU: http://www.pku.es/
Si tienes estudios en Ciencias de la Salud y quieres profundizar más en el diagnóstico y tratamiento de Enfermedades Raras, no te puedes perder nuestro Programa de Experto en Genética Clínica y Enfermedades Raras en colaboración con la Universidad Politécnica de Valencia.


