INTRODUCCIÓN
El trastorno por consumo de alcohol (TCA) es un trastorno crónico recurrente que progresa a través de un ciclo de adicción de tres etapas en el que intervienen circuitos neuronales de los ganglios basales, la amígdala extendida y el córtex prefrontal (Koob y Volkow, 2010). A lo largo de las décadas se han utilizado diferentes terminologías y enfoques diagnósticos para el TCA y los rasgos relacionados, incluyendo la dependencia del alcohol y el abuso de alcohol basados en el DSM-IV (American Psychiatric Association, 1994) y ediciones anteriores, y el TCA basado en el DSM-5 (American Psychiatric Association, 2013). Los criterios diagnósticos incluyen la tolerancia a los efectos del alcohol, el síndrome de abstinencia en ausencia de consumo de alcohol, la incapacidad para controlar o reducir la ingesta de alcohol, la preocupación por el alcohol en detrimento de las prioridades laborales, familiares y sociales, y otros (Kranzler y Soyka, 2018). Además, los códigos diagnósticos de la Clasificación Internacional de Enfermedades se utilizan ampliamente en los entornos clínicos y se reflejan en las historias clínicas electrónicas para diagnosticar el TCA y los trastornos relacionados. Si no se especifica lo contrario, en lo sucesivo utilizaremos una definición amplia de TCA para englobar tanto la dependencia del alcohol como el trastorno por consumo de alcohol.
El TCA y el consumo excesivo de alcohol contribuyen en gran medida a la carga mundial de morbilidad y causan importantes efectos adversos para la salud (GBD 2016 Alcohol Collaborators, 2018). Sin embargo, sólo tres medicamentos (disulfiram, naltrexona y acamprosato) están aprobados por la FDA estadounidense para el tratamiento del TCA (Kranzler y Soyka, 2018). Así pues, persiste la falta de opciones terapéuticas a pesar de la importancia clínica del problema.
GENÉTICA DEL TRASTORNO POR CONSUMO DE ALCOHOL: GENES CANDIDATOS Y ESTUDIOS DE LIGAMIENTO
El TCA es un trastorno complejo con importantes componentes ambientales y genéticos. Las influencias genéticas en el TCA están establecidas desde hace tiempo (Smith et al., 1971; Boston y Li, 1981, Pickens et al., 1991), y los estudios familiares y de gemelos han reportado una heredabilidad genética de aproximadamente 0,50 (IC 95%, 0,43-0,53) (Agarwal et al., 1981; Kendler et al., 1994; Reed et al., 1996; Heath et al., 1997; Verhulst et al., 2015). Tres oleadas de estudios genéticos realizados en las últimas décadas han identificado genes de susceptibilidad (Dick y Bierut, 2006; Gelernter y Kranzler, 2009; Edenberg y Foroud, 2013; Tawa et al., 2016, Deak et al., 2019). La primera oleada incluyó estudios de genes candidatos. En esta revisión, omitimos de la discusión los estudios con poco poder estadístico. Los genes candidatos relacionados con el metabolismo del etanol se investigaron intensamente en busca de asociaciones con el TCA (Thomasson et al., 1991; Borras et al., 2000; Chambers et al., 2002; Whitfield, 2002; Edenberg, 2007; Peng et al., 2014; Peng et al., 2017).
Las alcohol deshidrogenasas (ADH), como ADH1B y ALDH2, son enzimas que oxidan el etanol en acetaldehído, y las aldehído deshidrogenasas son enzimas que catalizan los aldehídos en sus ácidos correspondientes. La importancia de variantes funcionales como rs1229984 (que codifica His48Arg) en el gen ADH1B, y rs671 (que codifica Glu504Lys) en ALDH2 está bien establecida. La mayor eficacia catalítica de ADH1B (conferida por el alelo His48) o la menor actividad de ALDH2 (por el alelo Lys504, que es común exclusivamente en los asiáticos orientales) conduce a la acumulación de acetaldehído y al rubor (Mizoi et al., 1979; Crabb et al., 1989), lo que disuade de seguir ingiriendo alcohol, protegiendo así contra el TCA (Harada et al., 1982; Goedde et al., 1983; Li et al., 2012; Bierut et al., 2012). Otra variante codificante, ADH1B*rs2066702 (Arg369Cys), se ha asociado con el TCA, pero sólo en poblaciones africanas (a menos que se especifique lo contrario, las muestras de ascendencia africana en los estudios mencionados en este artículo eran afroamericanas), ya que no es polimórfica en otras poblaciones (Edenberg et al., 2006; Ehlers et al., 2001). Numerosos genes candidatos adicionales no han sobrevivido a la era de los GWAS (Estudios de Asociación del Genoma Gompleto), y no se tratarán aquí.
La segunda oleada de estudios genéticos incluyó estudios de ligamiento, que utilizan datos familiares para identificar regiones genómicas asociadas con el TCA (Long et al., 1998; Reich et al., 1998). A esta oleada le siguieron estudios posicionales de genes candidatos para localizar los genes o variantes relevantes. Se han identificado varios genes candidatos mediante análisis de ligamiento, incluidos genes que codifican receptores GABA (GABRA1 y GABRA2), (Covault et al., 2004; Edenberg et al., 2004; Enoch et al., 2009; Ittiwut et al., 2012), CHRM2 (receptor colinérgico muscarínico 2) (Wang et al., 2004; Luo et al., 2005) y otros. Sin embargo, estos genes no se identificaron en GWAS posteriores.
ESTUDIOS DE ASOCIACIÓN DEL GENOMA COMPLETO EN TRASTORNO POR CONSUMO DE ALCOHOL
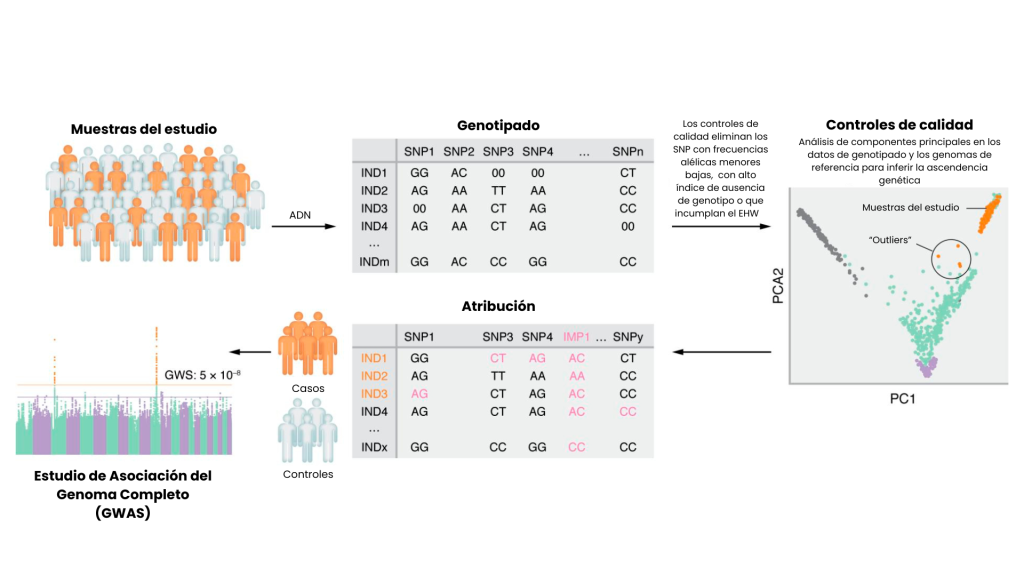
A diferencia de los anteriores estudios de genes candidatos, el GWAS es un método libre de hipótesis que explora las variantes comunes en todo el genoma utilizando genotipado mediante microarrays o secuenciación para identificar asociaciones con los rasgos de estudio (Figura 1) (Klein et al., 2005; McCarthy et al., 2008). Se han realizado progresos sustanciales en la oleada de estudios genéticos del TCA mediante GWAS (Figura 2 y Tabla 1).
En 2009, se llevó a cabo el primer GWAS del TCA en una muestra alemana compuesta por 487 casos de TCA y 1358 controles basados en la población. Ninguna variante alcanzó el umbral de significación en todo el genoma (GWS) (Treitlein et al., 2009). En 2011, el mismo equipo aumentó el tamaño de la muestra reclutando a más participantes e identificó una variante localizada entre ADH1B y ADH1C. En este estudio, se investigó por primera vez la puntuación de riesgo poligénico (PRS, polygenic risk score, un método que cuantifica la predisposición genética de un individuo a un rasgo o enfermedad particular sumando los efectos de múltiples variantes genéticas a través del genoma) para el TCA con el fin de probar la asociación con el TCA en muestras independientes, incluyendo la división aleatoria de las muestras del estudio en dos grupos, más dos muestras de la Base de Datos de Genotipos y Fenotipos de la Biblioteca Nacional de Medicina (Frank et al., 2012). En 2010, un estudio de muestras holandesas y australianas fue el primer GWAS de TCA que aplicó la imputación de SNP ausentes utilizando el panel de referencia HapMap (International HapMap C, 2003). El estudio no proporcionó resultados de GWAS para el TCA, pero se identificaron tres SNP para el TCA comórbido y la dependencia de la nicotina (Lind et al., 2010). No se identificó ninguna asociación en una muestra comunitaria general en Australia, pero este estudio discutió la naturaleza poligénica del TCA y proyectó la necesidad de un mayor tamaño de la muestra (Heath et al., 2011). Se realizaron nuevos análisis de estas cohortes para aumentar el poder estadístico (Wang et al., 2011; Zuo et al., 2012; Wang et al., 2013; Wetherill et al., 2014).
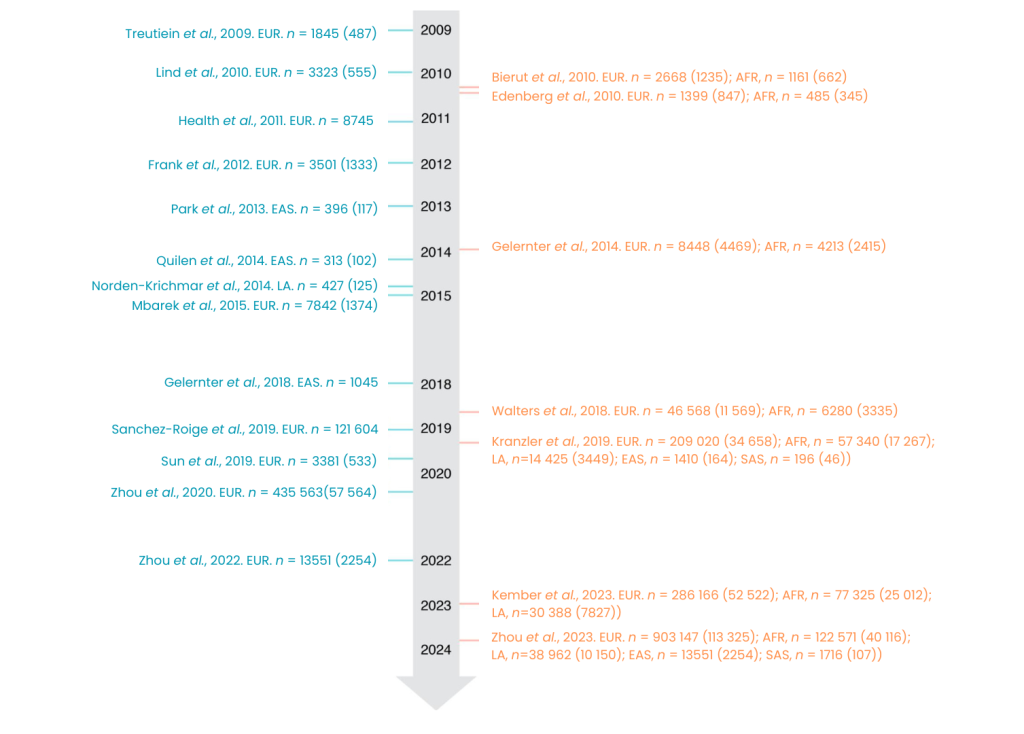
El grado insuficiente de diversidad genética en las poblaciones de estudio ha sido un reto persistente en los estudios de genética humana, siendo la mayoría de los participantes en los estudios de ascendencia europea (Peterson et al., 2019; Sirugo et al., 2019). La inclusión de poblaciones no europeas en los GWAS del TCA podría ayudar a esclarecer las arquitecturas genéticas compartidas y específicas entre poblaciones. Tres GWAS del TCA ampliaron el esfuerzo de descubrimiento de genes a más poblaciones (Bierut et al., 2010; Edenberg et al., 2010; Norden-Krichmar et al., 2014). Sin embargo, en estos estudios no se identificaron señales de GWAS. Posteriormente, se realizaron varios GWAS del TCA en muestras de Asia oriental. El primero fue un estudio de una muestra coreana con 396 individuos no emparentados, que identificó tanto el ADH1B*rs1229984 como el ALDH2*rs671 (Park et al., 2013). Hasta ahora, las conocidas variantes funcionales codificantes rs1229984 y rs671 específicas de Asia oriental han sido confirmadas por el enfoque GWAS. Otros estudios también identificaron que la región ALDH2 está asociada con el TCA en muestras de Asia Oriental (Quillen et al., 2014; Gelernter et al., 2018; Sun et al., 2019; Zhou et al., 2022), sin que se hayan identificado variantes de riesgo adicionales más allá de estas dos regiones.
El metaanálisis (un método que combina resultados de GWAS de dos o más cohortes separadas) de muestras recién reclutadas con datos resumidos publicados previamente ofrece la oportunidad de descubrir variantes de riesgo adicionales. En 2014, se llevó a cabo un estudio en el que participaron más de 10 000 individuos de ascendencia africana y europea, combinando varias cohortes. Tanto en el análisis de casos y controles como en el de criterios, se confirmó la región del gen ADH y se identificó una fuerte asociación con la variante codificante rs2066702 (Arg369Cys) en ADH1B en muestras africanas. Otros cuatro loci se asociaron con el TCA en el análisis de recuento de criterios: dos en muestras de ascendencia europea y dos en muestras de ascendencia africana (Gelernter et al., 2014). Un gran metaanálisis del TCA del Consorcio de Genómica Psiquiátrica combinó 28 estudios de individuos de ascendencia tanto europea (n = 46 568) como africana (n = 6280), confirmando las asociaciones con el grupo de genes ADH; sin embargo, no se descubrieron variantes de riesgo adicionales (Walters et al., 2018). Este estudio también investigó las correlaciones genéticas entre el TCA y muchos otros rasgos, observando correlaciones significativas con trastornos psiquiátricos, rasgos de consumo de sustancias y estatus socioeconómico (nivel educativo y puntuación de privación de Townsend). Los PRS derivados de los GWAS europeos mostraron predicciones más débiles en la muestra africana independiente que los PRS derivados de los GWAS africanos, lo que indica una capacidad limitada de los PRS entre poblaciones de diferente origen ancestral (Martin et al., 2017).
Además del diagnóstico basado en DSM o la Clasificación Internacional de Enfermedades, el TCA puede evaluarse mediante el Test de Identificación de Trastornos por Consumo de Alcohol (AUDIT, Alcohol Use Disorders Identification Test), un cuestionario de 10 preguntas desarrollado por la OMS para medir el consumo peligroso o perjudicial de alcohol en el último año (Saunders et al., 1993). Las preguntas 1-3 están dirigidas a evaluar los niveles de consumo de alcohol (AUDIT-C), y las preguntas 4-10 se centran en evaluar el consumo problemático de alcohol (AUDIT-P). El AUDIT es útil para detectar el consumo problemático de alcohol (Allen et al., 1997; Boschloo et al., 2010) por lo que este cuestionario podría implementarse como una estrategia efectiva para el fenotipado de muestras en cohortes a gran escala o biobancos.
Tabla 1. Estudios de asociación de genoma completo (GWAS) en trastorno por consumo de alcohol.
| Estudio | Detalles | Resultados | Notas |
| Treutlein et al., 2009 | EUR n = 1183 Casos = 487 | Sin resultados significativos a nivel genómico | Todo hombres |
| Lind et al., 2010 | EUR n = 1139 Casos = 847 | Sin resultados significativos a nivel genómico | Primera imputación |
| Edenberg et al., 2010 | AFR n = 1235 Casos = 1235 | Sin resultados significativos a nivel genómico | Score TCA |
| Bierut et al., 2010 | EUR n = 2686 Nc = 1035 | Sin resultados significativos a nivel genómico | |
| Health et al., 2011 | EUR n = 8754 | Sin resultados significativos a nivel genómico | Todo hombres |
| Frank et al., 2012 | EAS n = 1333 Casos = 1333 | rs1798891 | Todo hombres |
| Park et al., 2013 | EAS n = 396 Casos = 117 | ADH1Brs1229984 y ALDH2rs671 | |
| Gelernter et al., 2014 | AFR n = 4629 | ADH1Brs1229984 ADH1Brs2066702 en AFR y 4 otros loci | Rasgo ordinal |
| Quillen et al., 2014 | EUR n = 8448 Casos = 4143 | ADH1Brs1229984 ADH1Brs2066702 en AFR | Case-control |
| Norden-Krichmar et al., 2014 | EAS n = 427 Casos = 425 | ALDH2*rs671 | Todo hombres |
| Mbarek et al., 2015 | EUR n = 7842 Casos = 1374 | Sin resultados significativos a nivel genómico | AUDIT case/control |
| Gelernter et al., 2018 | EAS n = 2123 Casos= 1266 | ALDH2 locus | Rasgo ordinal |
| Walters et al., 2018 | EUR n = 46568 Casos = 11559 AFR n = 6,280 Casos = 3335 | ADH1Brs1229984 ADH1Brs2066702 en AFR | |
| Sanchez-Roige et al., 2019 | EUR n = 20328 | Sin resultados significativos a nivel genómico | AUDIT |
| Sanchez-Roige et al., 2019 | EUR n = 141932 | 15 variantes independientes en 11 loci para AUDIT; KLB, región ADH, SLC39A8 paraAUDIT-P | AUDIT, AUDIT-P |
| Kranzler et al., 2019 | EUR n = 209020 Casos= 34658 AFR n = 57340 Casos= 17267 LA n = 14245 Casos = 3449 EAS n = 1410 Casos = 164 SAS n = 196 Casos = 46 | 10 loci en EUR; 2 en AFR; 2 en LA; 15 variantes independientes en todos los análisis | |
| Sun et al., 2019 | EAS n = 3181 Casos = 533 | región ADH y ALDH2 | Todo hombres |
| Zhou et al., 2020 | EUR n = 435563 Casos = 57564 | 24 variantes independientes en TCA; 29 con Consumo problemático de alcohol | Consumo problemático de alcohol |
| Zhou et al., 2022 | EAS n = 13551 Casos = 2254 | región ADH y ALDH2 | |
| Kember et al., 2023 | EUR n = 286166 Casos = 52522 AFR n = 77325 Casos = 25012 LA n = 30,388 Casos = 7827 | 19 variantes independientes en EUR; 4 en AFR; 1 en LA; 32 en todos los análisis | También se probó una definición de TCA menos estricta |
| Zhou et al., 2023 | EUR n = 903147 Casos = 113325 AFR n = 122571 Casos = 40116 LA n = 38962 Casos = 10150 EAS n = 13551 Casos = 2254 SAS n = 1716 Casos = 107 | 85 variantes independientes en EUR; ADH1Brs1229984 y rs2066702 en AFR; ADH1Brs1229984 en LA; región ADH y ALDH2 en EAS; 110 en todos los análisis | Consumo problemático de alcohol |
EUR: población europea; AFR: población africana; EAS: población asiática del este; LA: población latinoamericana; SAS: población asiática del sur; ADH: alcohol deshidrogenasa; AUDIT: Prueba de Identificación de Trastornos por Consumo de Alcohol; Nc: número de casos.
a Solo se incluyen aquí los estudios con nuevas muestras. En esta tabla se presentan los resultados significativos a nivel genómico de las muestras de descubrimiento, sin incluir algunos resultados combinados con muestras de replicación.
b Después de análisis condicionales.
c No se realizaron análisis condicionales en cada ascendencia; este número es un valor aproximado.
d Señales independientes en el metaanálisis entre ascendencias más las señales específicas de cada grupo de ascendencia.
Los dos primeros GWAS de las puntuaciones del AUDIT no identificaron ninguna asociación (Sanchez-Roige et al., 2019; Mbarek et al., 2015). Un estudio posterior del AUDIT en dos cohortes de base poblacional, el Biobanco del Reino Unido (Bycroft et al., 2018) y 23andMe (Sangez-Roige et al., 2019), con un total de 141 932 participantes, identificó 15 señales independientes en 11 loci genómicos para la puntuación total del AUDIT, muchas de ellas novedosas (Sanchez-roige et al., 2019). Cuatro loci se asociaron con la subpuntuación AUDIT-P, incluida la región ADH, KLB (que codifica β-klotho) y SLC39A8 (familia de transportadores de solutos 39 [transportador de zinc], miembro 8). Otro hallazgo clave de este estudio es que la arquitectura genética del AUDIT-P difiere de la del AUDIT-C, y que el AUDIT-P está genéticamente más correlacionado con el TCA que el AUDIT-C. El estudio de asociación de todo el transcriptoma (TWAS) (Barbeira et al., 2018) identificó 26 genes cuya expresión génica prevista en los tejidos cerebrales se asociaba con el AUDIT.
Un estudio del Programa del Millón de Veteranos (MVP) (Gaziano et al., 2016) investigó tanto el TCA como el consumo de alcohol (medido con el AUDIT-C) en cinco grupos de población, incluidas poblaciones europeas, africanas, de Asia oriental, latinoamericanas y del sur de Asia (Kranzler et al., 2019). Este estudio incluyó 274 391 participantes, con 55 584 diagnosticados de TCA según los códigos de la Clasificación Internacional de Enfermedades. Se identificaron 15 variantes independientes (tras análisis condicionales) en 10 loci en múltiples ascendencias, incluidas 10 en ascendencias europeas, 2 en ascendencias africanas y 2 en ascendencias latinoamericanas. El análisis de heredabilidad por particiones para investigar cómo las categorías funcionales específicas del tipo celular del genoma contribuyen a la heredabilidad de una enfermedad compleja (Finucane et al., 2015) indicó que el grupo de tipos celulares más significativamente enriquecido para el TCA es el correspondiente al sistema nervioso central, confirmando con evidencias genéticas que el TCA es un trastorno relacionado con el cerebro. Este estudio también aportó el resultado clave de que la arquitectura genética del consumo de alcohol (medida por el AUDIT-C) difiere de la del TCA (se observó un patrón similar entre el AUDIT-C y el AUDIT-P, ref. Sanchez-Roige et al., 2019), subrayando que analizar el TCA o el AUDIT-P por separado de los rasgos de consumo de alcohol reduciría la heterogeneidad.
Antes de estos dos artículos clave, no se había reconocido que las medidas de cantidad o frecuencia difieren genéticamente (y por tanto biológicamente) respecto a las de dependencia. Otro estudio de diferentes ascendencias utilizó datos longitudinales del Programa del Millón de Veteranos y confirmó esta diferencia entre el TCA y AUDIT-C e identificó nuevos loci con ambos rasgos. En concreto, este estudio identificó un conjunto de variantes con efectos sobre el TCA que no están mediados por el consumo de alcohol (es decir, AUDIT-C) (Kember et al., 2023).
Un estudio posterior sobre el consumo problemático de alcohol, un fenotipo indirecto del TCA, combinó los datos de TCA del Programa del Millón de Veteranos y el Consorcio de Genómica Psiquiátrica y el AUDIT-P del UK Biobank e identificó 29 variantes de riesgo independientes en 435 563 participantes EUR (Zhou et al., 2020). En este estudio, la correlación genética entre el AUDIT-P y el TCA se estimó en 0,71 (error estándar = 0,05), lo que justifica el metaanálisis del fenotipo indirecto del TCA en estos conjuntos de datos. Este estudio señaló la heterogeneidad entre estos fenotipos y discutió que las asociaciones específicas de cada definición podrían haberse atenuado. Se encontró un total de 327 interacciones conocidas entre fármacos y genes para 16 genes asociados, siendo DRD2 el que más interacciones farmacológicas presentaba (n = 177), seguido de BDNF (n = 68) y PDE4B (n = 36).
El análisis de valor de riesgo poligénico de todo el fenoma en el biobanco independiente BioVU confirmó las correlaciones genéticas entre el consumo problemático de alcohol y el consumo de sustancias y los trastornos psiquiátricos. Las vías que incluyen la oxidación del etanol en el reactoma y el metabolismo del etanol y el alcohol fueron las más significativamente enriquecidas para el TCA. Los TWAS mostraron enriquecimientos significativos en varios tejidos cerebrales, incluidos el cerebelo y el córtex, lo que ilustra aún más los mecanismos específicos de tejido de esta enfermedad relacionada con el cerebro. El análisis de aleatorización mendeliana (Sanderson et al., 2022), un conjunto de métodos que utiliza variantes genéticas como variables instrumentales para estimar la relación causal entre la exposición y el resultado, sugirió que la responsabilidad ante el consumo de sustancias, el estado psiquiátrico, la conducta de riesgo y el rendimiento cognitivo tienen efectos causales sobre la responsabilidad ante el consumo problemático de alcohol.
RESULTADOS DEL GWAS DEL CONSUMO PROBLEMÁTICO DE ALCOHOL CON MÚLTIPLES ASCENDENCIAS REALIZADO EN 2023
Hasta ahora, los estudios han identificado genes de riesgo asociados con el consumo problemático de alcohol en múltiples ascendencias y han confirmado repetidamente las asociaciones de varios genes, principalmente en poblaciones con ascendencia europea.
En 2023, un estudio multiancestral de consumo problemático de alcohol con más de 1 millón de participantes reveló numerosos hallazgos novedosos (Zhou et al., 2023). Se identificaron 85 variantes de riesgo independientes en participantes con ascendencia europea, y 110 variantes de riesgo en total en metaanálisis dentro de una misma ascendencia o entre ascendencias. El mapeo cruzado entre ancestros identificó conjuntos creíbles en 13 loci (un conjunto de variantes causales plausibles dentro de cada locus; estos conjuntos de variantes causales putativas se denominan “conjuntos creíbles”) que contenían una única variante. Había 34 conjuntos creíbles adicionales que contenían entre 2 y 5 variantes. En conjunto, estos resultados proporcionaron una lista de variantes diana para futuros estudios funcionales experimentales. Aprovechando la información de múltiples ascendencias, los resultados de la asociación de valores de riesgo poligénico entre ascendencias fueron mayores que los obtenidos con valores de riesgo poligénico de una sola ascendencia (Ruan et al., 2022). En este estudio se examinaron los genes solapados tanto mediante análisis de asociación génica como mediante TWAS (Barbeira et al., 2019) en tejidos cerebrales y/o análisis de interacción de la cromatina (Sey et al., 2020) utilizando anotaciones cerebrales Hi-C (Captura de Conformación Cromosómica de Alta Capacidad, en sus siglas en inglés). Muchos genes mostraron pruebas convergentes que vinculaban la asociación al consumo problemático de alcohol con la biología cerebral mediante análisis de expresión génica (TWAS) y de interacción de la cromatina (Hi-C). Traducir los resultados genéticos en aplicaciones clínicas es un objetivo importante de los estudios genéticos humanos, y estudios previos han demostrado las posibilidades (Nelson et al., 2015; King et al., 2019; Backman et al., 2021). A través de dos tipos de análisis de reutilización de fármacos, este estudio identificó medicamentos existentes como tratamientos potenciales para el TCA. En el primer análisis se buscaron las señales genéticas independientes en Open Targets (Ochoa et al., 2023) para determinar si eran susceptibles de convertirse en fármacos y si eran objetivos de medicación. Muchos genes eran susceptibles de ser tratados, como DRD2, CACNA1C, DPYD, PDE4B, KLB, BRD3, NCAM1, FTO, MAPT, OPRM1 y GABRA4. El segundo análisis de reutilización de fármacos, utilizando los resultados de TWAS, halló que 287 compuestos estaban significativamente correlacionados con el patrón transcripcional asociado al riesgo de TCA.
Entre estos compuestos se encuentran la tricostatina-a, la melperona, la triflupromazina, la espironolactona, el amlodipino y el clometiazol. La tricostatina-a tiene efectos en la prevención del desarrollo de la ansiedad relacionada con la abstinencia de alcohol en ratas (Pandey et al., 2008), el clometiazol se utiliza para tratar el síndrome de abstinencia de alcohol (Sychla et al., 2017), y la espironolactona reduce el consumo de alcohol tanto en ratas como en humanos con pruebas convergentes (Farokhnia et al., 2022). Este estudio proporcionó una lista de posibles medicamentos y dianas para futuros estudios farmacológicos del TCA.
LIMITACIONES DE LOS ESTUDIOS DE ASOCIACIÓN DEL GENOMA COMPLETO EN TRASTORNO POR CONSUMO DE ALCOHOL Y FUTURAS DIRECCIONES
Aunque el campo de la genética del TCA ha progresado considerablemente, persisten lagunas sustanciales (similares a las de otros trastornos psiquiátricos, Derks et al., 2022).
A continuación, destacamos algunas limitaciones de los estudios actuales sobre el TCA (Tabla 2) con la esperanza de que las lagunas puedan llenarse con nuevos conjuntos de datos, tecnologías, métodos analíticos y direcciones de investigación en el futuro.
Tabla 2. Desafíos de los estudios genéticos del trastorno por consumo de alcohol.
| Desafío | Explicación | Soluciones |
| 1. Fenotipado profundo | Heterogeneidad debido a diferentes definiciones | Usar una misma definición para reducir la heterogeneidad y centrarse en subfenotipos para identificar variantes específicas |
| 2. Diversidad genética | La mayoría de las muestras estudiadas son de ascendencia europea | Reclutar individuos de diversas ascendencias genéticas, por ejemplo, mediante biobancos globales o reclutamiento dirigido de poblaciones no europeas |
| 3. Rasgo poligénico | Cientos de miles de variantes contribuyen al riesgo con efectos pequeños | Aumentar el tamaño de la muestra; integrar datos de biobancos |
| 4. Genoma incompleto | Falta de información sobre variantes raras y variantes estructurales | La secuenciación del genoma completo podría extender el descubrimiento de genes a todo el genoma |
| 5. Funciones | La mayoría de las variantes identificadas están en regiones no codificantes con funciones desconocidas | Usar métodos de aprendizaje profundo para predecir funciones de variantes; validar con ensayos experimentales |
| 6. Vías biológicas | No se conocen las vías biológicas | Utilizar nuevos conjuntos de datos (epigenética, multi-ómica) y tecnologías (secuenciación de célula única, transcriptómica espacial) para investigar los mecanismos |
| 7. Interferencia por factores de confusión | Los efectos genéticos indirectos y factores de confusión pueden sesgar los resultados | Separar los efectos genéticos directos de los indirectos y considerar los factores de confusión en el análisis |
| 8. Predicción | Bajo rendimiento en la predicción del riesgo basada en valores de riesgo poligénico | Aprovechar el big data y la inteligencia artificial, quizás en un futuro próximo |
(a) Diferentes definiciones de TCA y fenotipos sustitutos (p. ej., AUDIT-P) tienen una arquitectura genética compartida, lo que resulta en una mayor potencia en el descubrimiento de genes cuando se combinan a partir de diferentes cohortes (Zhou et al., 2020; Sanderson et al., 2022; Zhou et al., 2023). Sin embargo, no son rasgos idénticos. El fenotipado profundo (ya sea utilizando la misma definición o centrándose en subfenotipos) en cohortes más grandes podría reducir la heterogeneidad fenotípica y aumentar la posibilidad de identificar asociaciones y vías específicas de rasgos (Cai et al., 2020).
(b) Algunos estudios se han esforzado por incluir muestras de múltiples ascendencias (Bierut et al., 2010; Edenberg et al., 2010; Gelernter et al., 2014; Walters et al., 2018; Kranzler et al., 2019; Zhou et al., 2023), pero el tamaño de las muestras de las ascendencias no europeas es menor que el de las muestras de las ascendencias europeas, un problema común en los estudios de genética humana (Peterson et al., 2019; Sirugo et al., 2019). El reclutamiento de individuos de diversas ascendencias genéticas es un paso crítico en este campo. Con la disponibilidad de más biobancos multiancestrales, como el Programa del Millón de Veteranos, la iniciativa Global Biobank Meta-analysis (Zhou et al., 2022) y el programa de investigación All of Us (All of Us Research Program Investigators, et al., 2019), prevemos que la brecha en la diversidad disminuirá. Los organismos de financiación también deberían prestar atención a los estudios que proponen un reclutamiento centrado en participantes de ascendencia no europea.
(c) El TCA es un trastorno altamente poligénico, con cientos de variantes que, como mínimo, contribuyen al riesgo (Zhou et al., 2023; Gelernter et al., 2021). La aproximación de GWAS de “fuerza bruta” requiere una muestra de mayor tamaño para identificar más variantes de riesgo. A diferencia de otros rasgos o comportamientos que pueden medirse directamente y evaluarse en grandes poblaciones o biobancos -por ejemplo, los GWAS de la estatura (Yengo et al., 2022), el nivel educativo (Okbay et al., 2022) y el consumo de alcohol (Saunders et al., 2022) se han realizado en 3~5 millones de participantes-, el diagnóstico clínico del TCA en grandes cohortes aún está retrasado. Al igual que en el punto (a), el aumento del tamaño de la muestra y la incorporación de múltiples ascendencias podrían mejorar la potencia y la resolución del mapeo fino de variantes causales (Zhou et al., 2023). Además de las conocidas variantes funcionales codificantes en los genes metabólicos del alcohol, la mayoría de las variantes identificadas a través de grandes GWAS tienen efectos pequeños o muy pequeños sobre el riesgo de TCA, lo que reduce el rendimiento del amplio esfuerzo de seguimiento de los estudios funcionales sobre variantes individuales. Este es un problema común en el estudio genético de rasgos complejos.
(d) Los estudios GWAS actuales han utilizado principalmente matrices de SNPs e imputación post hoc para rellenar las variantes comunes, lo que no permite el análisis del genoma completo ya que algunas partes del genoma no están totalmente “cubiertas”, es decir, hay variantes no evaluadas en algunas regiones genómicas que no pueden someterse a pruebas de asociación, por razones técnicas. Una matriz de SNPs típica puede capturar entre 600 000 variantes (por ejemplo, Illumina PsychArray) y 1,8 millones (por ejemplo, Illumina Multi-Ethnic Genotyping Array). Tras la imputación y la aplicación de controles de calidad estándar para las variantes, los números típicos analizables de variantes de alta calidad varían de 5 a 15 millones, dependiendo de la densidad de SNPs de la matriz original, el tamaño de la muestra y la ascendencia genética (desde el punto de vista de la genética de poblaciones, las poblaciones africanas tienen más variantes comunes que otras poblaciones debido a su historia evolutiva). Dada la información inherente que falta en los distintos pasos, los metaanálisis GWAS sólo pueden cubrir un subconjunto de variantes de todo el genoma, lo que indica que gran parte del genoma falta en los estudios genéticos actuales del TCA. La secuenciación de todo el genoma (WGS), que puede detectar esencialmente todas las variantes (incluidas las variantes raras y las variantes estructurales) sin sesgo de determinación, podría ofrecer mejores oportunidades para investigar la arquitectura genética completa del rasgo.
Recientemente se han llevado a cabo varios estudios de secuenciación del exoma completo (WES) y un estudio WGS del TCA (Wang et al., 2021; Gentry et al., 2023; Hill y Hostyk, 2023; Wang et al., 2024). Un estudio WES de todo el fenoma de 170 979 individuos (6320 casos) del Biobanco del Reino Unido identificó dos variantes comunes en el gen ADH1C asociadas con el TCA (P < 2 × 10-9), utilizando un modelo aditivo o dominante (Wang et al., 2021). Un estudio WES que combinó 469 835 individuos de los datos del Biobanco del Reino Unido (13 121 casos) y 3.789 individuos de la cohorte Yale-Penn (2562 casos) con múltiples ancestros identificó la conocida variante funcional ADH1B*rs1229984 y varias variantes comunes en ADH1C. Las pruebas basadas en genes que tienen en cuenta la carga de variantes de pérdida de función, sin sentido y sinónimas identificaron los nuevos genes CNST e IFIT5 (Wang et al., 2024). Un estudio de WGS de baja cobertura sobre acontecimientos vitales relacionados con el TCA y dos síntomas afectivos en 742 indios americanos y 1711 americanos europeos identificó nuevas variantes tanto comunes como raras (Penq et al., 2019).
(e) La mayoría de las variantes identificadas por GWAS se encuentran en regiones no codificantes con funciones desconocidas (Edwards et al., 2013). Las variantes más asociadas en cada locus de riesgo no son necesariamente las variantes causales del TCA. Aunque el análisis de mapeo fino posterior al GWAS podría identificar un conjunto creíble de posibles variantes causales (Welcome Trust Case Control Consortium et al., 2012; Huang et al., 2017; Yuan et al., 2017), se necesitan más esfuerzos para interpretar y validar las funciones de las variantes. En los últimos años, se han aplicado con éxito en la investigación biomédica enfoques analíticos novedosos como el aprendizaje profundo (un subconjunto del aprendizaje automático). Por ejemplo, los métodos de aprendizaje profundo contribuyen a la predicción de la estructura de las proteínas (Jumper et al., 2021; Lin et al., 2023), las variantes patógenas sin sentido (Cheng et al., 2023; Gao et al., 2023) y las funciones reguladoras de las variaciones genómicas (Zhou y Troyanskaya, 2015; Zhou et al., 2018). La combinación de nuevas herramientas computacionales y ensayos funcionales de vanguardia como la edición del genoma (Morris et al., 2023; Gasperini et al., 2019; Wunnemann et al., 2023) podría ayudar a evaluar los efectos de las variantes a escala.
(f) Aunque se han identificado cientos de variantes de riesgo y muchas se han replicado repetidamente en GWAS, los efectos genéticos indirectos (también llamados “crianza genética”), que son efectos de los alelos en los padres sobre la descendencia a través del entorno (Trejo y Domingue, 2018), no se han distinguido de los efectos genéticos directos sobre el TCA. Se han desarrollado métodos para imputar genotipos parentales utilizando datos familiares (Young et al., 2022), que podrían utilizarse para mejorar las estimaciones de los efectos genéticos directos sobre el TCA. Los efectos de confusión, incluido el estatus socioeconómico, también pueden sesgar los resultados. Por ejemplo, el nivel educativo influye en muchos rasgos psiquiátricos y no psiquiátricos (Okbay et al., 2022) y tiene una correlación genética rg = -0,21 con el TCA, que debe tenerse en cuenta en futuros estudios.
(g) Otra profunda laguna es que el actual rendimiento predictivo de los valores de riesgo poligénicos para el TCA, basados en variantes comunes de GWAS (por ejemplo, utilizando la variación genética para predecir el riesgo en individuos genotipados) es fuertemente significativo a nivel estadístico, pero numéricamente es todavía débil y aún no ha entrado en el rango de utilidad clínica. A pesar del aumento del tamaño de la muestra, la heredabilidad basada en SNPs (h2) por GWAS es baja (la h2 oscila entre el 5,6% y el 12,7% mientras que la h2 a nivel de escala de predisposición oscila entre el 8,9% y el 16,2%, refs. Walters et al., 2018; Sanchez-Roige et al., 2019; Kranzler et al., 2019; Zhou et al., 2020; Zhou et al., 2023) en comparación con la heredabilidad total, pero comparable a la observada en muchos otros rasgos genéticamente complejos.
Actualmente, los valores de riesgo poligénico tienen un poder limitado para la predicción del TCA (varianza explicada medida por pseudo R2) en cohortes independientes; por lo tanto, el uso clínico de los valores de riesgo poligénico actuales para el TCA no es inminente. Posiblemente, el éxito de la inteligencia artificial en otras áreas podría extenderse a la predicción del riesgo de TCA, con más datos genómicos y de historias clínicas electrónicas a gran escala disponibles mediante la integración de datos genómicos mejorados con otros predictores de rasgos.
(h) Por último, los estudios genéticos han confirmado que el TCA es en parte un trastorno relacionado con el cerebro (Kranzler et al., 2019). Se ha dado prioridad a los genes con perturbación de la expresión en tejidos cerebrales específicos (Sanchez-Roige et al., 2019; Zhou et al., 2023), pero las vías biológicas que van de la genética a la etiología del TCA todavía no están claras en gran medida. Sin embargo, hay excepciones importantes: se conoce bien el mecanismo del efecto sobre el riesgo de la variación de las enzimas que metabolizan el alcohol. Muchos procesos biológicos desempeñan papeles en las vías, como la expresión génica, la regulación funcional, la perturbación de proteínas, los metabolitos y otros rasgos mediadores. Para comprender los mecanismos de las rutas biológicas, se necesitan estudios que vayan más allá de la genética y que incluyan, entre otros, la epigenética (analizada en el apartado Epigenética del TCA), la multiómica, la secuenciación unicelular y la transcriptómica espacial más reciente.
EPIGENÉTICA DEL TRASTORNO POR CONSUMO DE ALCOHOL
Los estudios epigenéticos del TCA han surgido como una vía importante para comprender la compleja interacción entre la genética, el entorno y la regulación génica en el desarrollo y la progresión del TCA. Los factores epigenéticos incluyen factores de transcripción, ARN no codificantes, modificaciones del ADN o modificaciones de las histonas que alteran la expresión génica y, en consecuencia, afectan a los fenotipos, sin cambiar la secuencia del ADN (Feil y Fraga, 2012; Holliday, 1987). Mientras que el estado epigenético es altamente heredable y se ve afectado por factores ambientales, incluyendo la exposición al alcohol, ciertos cambios epigenéticos en regiones cerebrales específicas han sido implicados en la etiología del TCA (Robinson y Nestler, 2011).
Aunque la mayoría de los estudios epigenéticos en humanos se han centrado en el consumo de alcohol (que no es el objetivo principal de esta Revisión; aquí nos centramos en el trastorno por consumo más que en el consumo), algunos estudios han explorado los patrones de metilación del ADN en individuos con TCA y han identificado metilación diferencial en regiones genómicas específicas (revisadas en las refs. Krishnan et al., 2014; Zhang y Gelernter, 2017; Longley et al., 2021; Nieratschker et al., 2013; Berkel y Pandey, 2018). Estos cambios se observan a menudo en genes relacionados con procesos neurobiológicos, sistemas de neurotransmisores y respuestas inmunes. Por ejemplo, se notificó una metilación del ADN significativamente mayor en el promotor de HERP en pacientes con TCA que en controles (Bleich et al., 2006), mientras que se observó un mayor nivel de metilación del ADN en la región promotora del gen OPRM1 en TCA (Zhang et al., 2012). Un estudio de cerebros humanos post mortem halló una disminución general de la metilación en los retrotransposones de repetición largo-terminal en la corteza frontal (Ponomarev et al., 2012). Cabe destacar que estas muestras de cerebro, junto con las utilizadas en varios estudios epigenéticos de seguimiento del TCA, procedían en su mayoría del Centro de Recursos de Tejidos de Nueva Gales del Sur (NSW TRC) de la Universidad de Sídney (Harper et al., 2003). Sin embargo, no se observaron diferencias globales de metilación entre los casos de TCA y los controles en el córtex frontal (Manzardo et al., 2012). También se notificó hipermetilación del ADN en otros genes, como SNCA (Bonsch et al., 2005), MAOA (Philibert et al., 2008), DAT (Hillemacher et al., 2009), NGF (Heberlein et al., 2013), AVP (Hillemacher et al., 2009), PDNY (Taqi et al., 2011) y GABRD (Gatta et al., 2017).
En un estudio de 285 afroamericanos y 249 europeos que utilizó una matriz de metilación diseñada a medida de 384 CpG en 82 genes candidatos, se identificó un sitio CpG significativo en la región promotora de HTR3A (receptor 3A de 5-hidroxitriptamina) en los europeos. También se notificaron otros CpG sugerentes en afroamericanos (en los genes GABRB3 y POMC) o en euroamericanos (en los genes NCAM1, DRD4, MBD3, HTR2B y GRIN1) (Zhang et al., 2013). Como en los estudios de variación genética, no está claro si los resultados de los “genes candidatos” se demostrarán estables a lo largo del tiempo en los estudios epigenéticos.
ESTUDIOS DE ASOCIACIÓN DE EPIGENOMA COMPLETO EN TRASTORNO POR CONSUMO DE ALCOHOL
La mayoría de los estudios de metilación del ADN en TCA se han centrado en regiones genéticas individuales y no han producido resultados replicables. Se necesitan exploraciones de todo el epigenoma para identificar a escala los cambios epigenéticos relacionados con el TCA. De forma similar a las oleadas de tecnologías en los estudios genómicos, se han aplicado microarrays y técnicas de secuenciación de nueva generación a los estudios epigenéticos del TCA (Tabla 3).
Tabla 3. Estudios de asociación de epigenoma completo en trastorno por consumo de alcohol.
| Estudio | Tejido | N | Array | Hallazgos | Notas |
| Zhao et al, 2013 | Sangre | EAS, n = 20 Casos = 10 | 450 000 | 865 sitios hipometilados 716 hipermetilados | Sitios significativos (con diferencia de expresión ≥20) |
| Zhang et al., 2013 | Sangre | EAS, n = 128 Casos = 63 | 27 000 | 1702 sitios hipometilados 8 hipermetilados | FDR < 0.005 |
| Philibert et al., 2014 | Sangre | (EUR)a,n = 66 Casos = 33 | 450 000 | 56 sitios CpG diferencialmente metilados | Ninguno se asocia con la abstinencia alcohólica |
| Weng et al., 2015 | Sangre | EAS, n = 10 Casos = 10b | 27 000 | No claro | Solo hombres |
| Ruggeri et al., 2015 | Sangre | EUR, n = 36 Casos = 18 | 385 000 | 77 sitios CpG, 68% hipermetilados | Parejas de gemelos monocigóticos |
| Wang et al., 2016 | Corteza prefrontal, (BA9) | EUR, n = 46 Casos = 23 | 450 000 | En hombres: 1201 sitios hipometilados, 611 hipermetilados | Solo análisis estratificados por sexo |
| Hagerty et al., 2016 | Precúneo | EURc, n = 96 Casos = 49 | 450 000 | 561 hipometilados 485 hipermetilados | |
| Lohoff et al., 2018 | Corteza prefrontal (BA9) | EUR, n = 46 Casos = 23 | 450 000 | 1 hipermetilado 2 hipometilados | |
| Gatta et al., 2021 | Corteza prefrontal (BA10) | EUR, n = 46 Casos = 23 | 850 000 | No claro | |
| Witt et al., 2020 | Sangre | EUR, n = 194 Casos = 99 | 850 000 | Abstinencia: 9,845 sitios CpG Recuperación: 6,094 sitios CpG | FDR < 0.05; Solo hombres |
| Lohoff et al., 2021 | Sangre | Población mixtad, n = 539 Casos = 336 | 850 000 | 5,101 sitios CpG | FDR < 0.05 |
| Meng et al., 2021 | PFC (BA9), NAc | EUR, n = 86 Casos = 39 | 450 000 | 2 regiones diferencialmente metiladas, sin sitios CpG individuales | Solo hombres; permutación P < 0.05 |
| Clark et al., 2022 | 4 tipos celulares (sangre); PFC | EUR, n = 1132 Casos = 323 | MBD-Seq | 3 sitios CpG en células T 1,397 en monocitos | FDR < 0.1 |
| Zillich et al., 2022 | ACC, PFC (BA9), CN, VS, PUT | EUR, n = 111 Casos = 53 | 850 000 | 2 sitios CpG en CN; 18 en VS | FDR < 0.05 |
| White et al., 2024 | PFC (BA9), NAc | EUR, n = 119 | 850 000 | 53 CpG en NAc; 31 en DL-PFC 105 en metaanálisis | FDR < 0.05 |
a Mayoría europeos, con algunas otras ascendencias; b Solo se compararon diferentes fases dentro de los casos; c Mismas muestras que en Wang et al., 2016
d 289 europeos americanos y 249 afroamericanos.
En el primer estudio de asociación de todo el epigenoma (EWAS) sobre el TCA se utilizaron muestras de sangre periférica de 10 casos de TCA y 10 controles de ascendencia asiática oriental. En total, se identificaron 865 sitios CpG hipometilados y 716 hipermetilados (definidos como puntuación de diferencia de expresión ≥20) (Zhao et al., 2013). El segundo estudio incluyó una cohorte de 128 varones de Asia oriental, con 63 individuos diagnosticados de TCA. En este estudio, se observaron niveles significativamente más bajos de metilación en los casos en comparación con los controles, con 1702 sitios hipometilados y 8 hipermetilados que alcanzaron FDR P < 0,005 (Zhang et al., 2013). Un estudio de 33 pacientes de centros de tratamiento del alcoholismo y 33 individuos que actuaron como controles sanos (sin consumir alcohol durante seis meses) -en su mayoría varones europeos- identificó 56 posiciones CpG metiladas diferencialmente. Ninguna de estas posiciones siguió siendo significativa tras el programa de tratamiento hospitalario de 30 días (Philibert et al., 2014). Un estudio longitudinal de muestras de Asia oriental identificó 149 genes hipermetilados y 51 hipometilados (P < 0,01) entre las fases sana (1990-1992) y dependiente (2003-2009) en 10 individuos (Weng et al., 2015). Dados los cambios en el estado de metilación con el cambio del estado de consumo de alcohol en los casos, se justifican más estudios sobre el TCA con un diseño experimental más robusto que elimine los efectos continuos del consumo de alcohol
Se han observado patrones similares en un estudio longitudinal más amplio de 99 casos de TCA hospitalizados y 95 individuos emparejados que actuaron como controles. En este caso se recogieron muestras de sangre de los participantes afectados en dos fases, una durante la abstinencia aguda de alcohol y otra tras dos semanas de recuperación en los centros de tratamiento. En comparación con los controles, se identificaron 9845 CpG durante la abstinencia alcohólica y 6094 después de dos semanas. La comparación de las dos fases dentro de los casos reveló 2876 sitios CpG metilados diferencialmente, lo que sugiere la reversibilidad de la metilación relacionada con el alcohol y la abstinencia (Witt et al., 2020). El estudio de asociación en epigenoma completo en 18 pares de gemelos monocigóticos discordantes, es decir, uno afectado y otro no en cada par, ha identificado 77 regiones metiladas diferencialmente en FDR < 0,05 (Ruggeri et al., 2015). Un estudio más amplio de TCA en 539 muestras de sangre informó de 5101 sitios CpG metilados diferencialmente significativos tras la corrección FDR. De éstos, 96 sitios CpG se replicaron en una segunda cohorte de 43 casos de TCA y 43 controles (Lohoff et al., 2018).
Los estudios mencionados anteriormente se realizaron principalmente con muestras de sangre, y las investigaciones posteriores se centraron en muestras cerebrales post mortem de NSW TRC para identificar cambios en la metilación del ADN asociados con el TCA. Un estudio analizó específicamente muestras de corteza prefrontal de 23 casos de TCA y 23 controles, con especial énfasis en análisis estratificados por sexo debido a una observación previa de un metiloma sesgado por sexo (Xu et al., 2014). Entre 32 varones (16 casos de TCA), se descubrieron 1201 sitios CpG hipermetilados y 611 hipometilados a un nivel FDR de 0,05; sin embargo, no se observaron resultados significativos en las mujeres (Wang et al., 2016). El reanálisis de los mismos datos, combinando ambos sexos, identificó tres sitios CpG tras la corrección basada en FDR (Lohoff et al., 2018). El estudio de asociación de epigenoma completo de 49 casos de TCA y 47 controles utilizando una matriz de 450 000 posiciones de metilación identificó 561 CpG hipometilados y se notificaron 485 CpG hipermetilados con P < 1 × 10-7 (Hagerty et al., 2016). Un pequeño estudio en muestras de cerebro de 23 casos de TCA y 23 controles, utilizando un array de metilación de mayor densidad (850 000 sitios CpG) no encontró loci con variantes significativas en el genoma (ver Figura 1C en ref. Gatta et al., 2021), aunque había 1218 CpGs con P nominal ≤ 0,001 (Gatta et al., 2021).
Varios estudios han investigado simultáneamente varias regiones cerebrales. Por ejemplo, un estudio de dos regiones cerebrales, incluyendo el córtex prefrontal y el núcleo accumbens en 86 individuos, no identificó sitios CpG individuales pero identificó dos regiones metiladas diferencialmente (permutación P < 0,05) mapeando las regiones aguas arriba de los genes ZFP57 y DLGAP2 (Meng et al., 2021). Un estudio de asociación en epigenoma completo en cinco regiones cerebrales de 111 individuos identificó dos sitios CpG metilados diferencialmente en la región del núcleo caudado y 18 en el estriado ventral, sin hallazgos significativos en las otras tres regiones (Zillich et al., 2022). A pesar de utilizar muestras de cerebro del mismo repositorio (NSW TRC), los hallazgos reportados en estos estudios fueron en gran medida incoherentes.
En un estudio adicional sobre el TCA, se analizaron muestras cerebrales de 119 individuos del Repositorio de Cerebros Humanos del Instituto Lieber para el Desarrollo Cerebral. Esta investigación identificó 53 CpGs asociados con el TCA en el núcleo accumbens y 31 en el córtex prefrontal dorsolateral (BA9) con un FDR P < 0,05, sin solapamiento entre las dos regiones. Los investigadores llevaron a cabo un meta-análisis en las dos regiones, revelando 21 CpGs adicionales, llevando el total a 105 CpGs únicos asociados al TCA en 120 genes (White et al., 2024). Comparando los resultados de este estudio con investigaciones previas sobre regiones cerebrales (Hagerty et al., 2016, Zillich et al., 2022, Clark et al., 2022), sólo un CpG intergénico, cg00402668 en la región cerebral BA9, alcanzó un nivel de significación de “búsqueda” (FDR P < 0,05) en la BA10 de Clark et al., pero no en otras regiones cerebrales. A nivel de genes, tres genes anotados en este estudio se solaparon con genes de Hagerty et al. (Hagerty et al., 2016). Al comprobar el solapamiento del 1% superior de los sitios CpG entre los estudios, se observó un enriquecimiento significativo para los resultados del núcleo accumbens en este estudio en los resultados del putamen y el estriado ventral de Zillich et al.
Los estudios entre tejidos han permitido avanzar en la comprensión de los mecanismos epigenéticos superpuestos. En un estudio de 1132 muestras de sangre compuestas por cuatro tipos de células y 50 muestras de cerebro, se evaluaron más de 21 millones de sitios CpG mediante secuenciación de dominios de unión a metil-CG. No se observaron asociaciones significativas en la sangre total ni en el cerebro. Sin embargo, para las células T y los monocitos, se identificaron 3 CpGs y 1397 CpGs con FDR P < 0,1, respectivamente. Un sitio CpG en el gen DLGAP1 se replicó significativamente, y otros 34 sitios se replicaron nominalmente en una muestra independiente de 73 casos de TCA y 339 controles. Además de la metilación del ADN, este estudio también evaluó la hidroximetilación de más de 26 millones de sitios CpG en las muestras cerebrales. Aunque ningún sitio individual alcanzó significación en todo el metiloma, el estudio observó un solapamiento significativo entre los sitios principales en EWAS específicos del tipo de célula sanguínea y EWAS tanto de metilación como de hidroximetilación en el cerebro. Esto representa la primera exploración de la metilación específica del tipo celular para el TCA en sangre y considera el papel de la hidroximetilación cerebral en el TCA (Clark et al., 2022).
OTROS ESTUDIOS EPIGENÉTICOS EN TRASTORNO POR CONSUMO DE ALCOHOL
Más allá de los estudios sobre la metilación del ADN en el TCA, se han realizado unos pocos estudios en el cerebro humano que consideran los ARN no codificantes y las modificaciones de las histonas. Los microARN son pequeños ARN no codificantes que regulan la expresión y/o traducción de los ARNm diana, y desempeñan un papel importante en diversos procesos biológicos (Filipowicz, et al., 2008). Un estudio de microARNs en la corteza prefrontal de 27 individuos identificó 12 microARNs regulados al alza (FDR P < 0,05) en casos de TCA (n = 14) comparados con controles (n = 13), lo que sugiere un papel regulador de los microARNs en la expresión génica del TCA (Lewohl et al., 2011). Un estudio de genes candidatos del ARN no codificante largo BDNF-AS en la amígdala humana implicó un efecto regulador sobre la expresión del gen BDNF en los casos de inicio temprano (antes de los 21 años) del TCA (n = 11) en comparación con los controles (n = 22), pero no en el TCA de inicio tardío (Bohnsack et al., 2019).
Un estudio del transcriptoma mediante RNA-Seq y de la trimetilación de la histona H3 lisina 4 (H3K4me3) mediante ChIP-Seq en muestras post mortem de hipocampo cerebral del Banco de Cerebros de la Universidad de Miami identificó 11 genes expresados diferencialmente con FDR P < 0,05 en los casos de TCA (n = 8) en comparación con los controles (n = 8). Sin embargo, ningún cambio en H3K4me3 alcanzó FDR P < 0,05 o se solapó con cambios de expresión (Zhou et al., 2011). El reanálisis de los datos utilizando enfoques de red identificó 7 módulos de coexpresión enriquecidos para los cambios asociados a H3K4me3 en los casos de TCA en comparación con los controles, lo que sugiere relaciones entre esta marca epigenética y la expresión génica (Farris et al., 2015). El pequeño tamaño de la muestra y la escasa potencia indican que estos resultados deben tomarse con cautela.
Otro estudio investigó la coexpresión génica y su relación con múltiples modificaciones epigenéticas para el TCA en tejidos cerebrales del NSW TRC. Se evaluó la expresión génica en el núcleo central y basolateral de la amígdala y en el córtex frontal superior de 17 casos de TCA y 15 controles, la metilación global de H3K4 en el córtex de 6 casos de TCA y 6 controles y la metilación del ADN de retrotransposones repetidos de terminación larga, y la histona H3K4me3 en el córtex de 5 casos de TCA y 5 controles (ChIP-Seq). Este estudio identificó componentes celulares críticos y determinantes epigenéticos previamente no reconocidos de las relaciones de coexpresión génica y descubrió nuevos marcadores de modificaciones de la cromatina en el cerebro humano (Ponomarev et al., 2012).
En conclusión, los estudios epigenéticos han aportado conocimientos limitados sobre los mecanismos moleculares subyacentes al TCA. Teniendo en cuenta la conocida complejidad genética y etiológica del riesgo de TCA, y las contribuciones tanto de los genes como del entorno, se necesitarán muestras más grandes para extraer conclusiones duraderas sobre la epigenética del TCA. La integración de la metilación del ADN, las modificaciones de las histonas y los ARN no codificantes en nuestra comprensión de la patogénesis del TCA es prometedora para identificar nuevas dianas terapéuticas y desarrollar intervenciones personalizadas. A medida que avanza la tecnología y se perfeccionan las metodologías de investigación, se espera que el campo de la epigenética contribuya profundamente a desentrañar las complejidades del TCA. Sin embargo, en la actualidad este campo se ve muy limitado por la falta de investigaciones bien fundamentadas.
LIMITACIONES DE LOS ESTUDIOS EPIGENÉTICOS EN EL TRASTORNO POR CONSUMO DE ALCOHOL
Hasta la fecha, los avances para desentrañar el panorama epigenético del TCA han sido muy limitados. La heterogeneidad del TCA, unida a los distintos grados de exposición al alcohol en los diferentes grupos y estadios del trastorno, introduce complejidades en la interpretación de los hallazgos, especialmente en muestras pequeñas. Además, la naturaleza dinámica de las modificaciones epigenéticas requiere diseños de estudio sofisticados para captar los cambios temporales a lo largo del desarrollo del TCA. Comparativamente, los estudios genéticos del TCA, principalmente a través de GWAS, han tenido tamaños de muestra mucho más grandes (muchos órdenes de magnitud), mientras que los estudios epigenéticos actuales, particularmente en el cerebro humano, son pequeños, y el estudio más grande hasta la fecha ha incluido a 119 individuos (White et al., 2024). Por último, la superposición de hallazgos entre tejidos y estudios es mínima (Longley et al., 2021, White et al., 2024). Aunque esto podría atribuirse a cambios epigenéticos específicos del tejido/contexto, también plantea la posibilidad de resultados falsos positivos.
En cuanto a los GWAS del TCA, serán necesarios estudios epigenéticos a mayor escala para generar hallazgos replicables y priorizar variaciones genómicas robustas para futuros estudios farmacológicos. Otras direcciones futuras que tienen el potencial de mejorar nuestra comprensión de los mecanismos epigenéticos del TCA incluyen las siguientes. En primer lugar, los estudios longitudinales de seguimiento de los cambios epigenéticos a lo largo del tiempo (Ng et al., 2012) podrían ayudarnos a comprender la relación temporal entre el consumo de alcohol y las modificaciones epigenéticas y a dilucidar la causa y el efecto en el TCA. En segundo lugar, la integración de los datos epigenéticos con otros datos ómicos (por ejemplo, la transcriptómica) debería ayudarnos a comprender mejor los mecanismos reguladores relevantes. En tercer lugar, es importante investigar cómo los factores ambientales (por ejemplo, el estrés y la dieta) interactúan con los factores epigenéticos para influir en el riesgo de TCA. En cuarto lugar, los estudios de asociación en epigenoma completo actuales se centran en los cambios a nivel tisular; la realización de análisis epigenéticos específicos para cada tipo de célula (por ejemplo, datos del epigenoma unicelular, ref. Guo et al., 2013) puede proporcionar una visión más profunda de los mecanismos moleculares.
CONCLUSIONES
Los estudios genéticos del TCA han hecho avanzar enormemente nuestra comprensión de su compleja etiología, mientras que los estudios epigenéticos han hecho progresos limitados. Aunque estos estudios han proporcionado información valiosa, siguen existiendo retos y lagunas en nuestra comprensión, lo que subraya la necesidad de seguir investigando y explorando, incorporando muestras más grandes con un fenotipado más profundo en poblaciones más diversas. La integración de conocimientos procedentes de estudios genéticos y epigenéticos es prometedora para el desarrollo de estrategias terapéuticas específicas y personalizadas, lo que representa un paso crucial para abordar la naturaleza polifacética del TCA.
La investigación futura debería tener como objetivo aumentar la potencia estadística, ampliar las poblaciones de estudio para abarcar diversos grupos y, de este modo, perfeccionar nuestra comprensión de los mecanismos implicados. La superación de las limitaciones y la traducción de los resultados de la investigación en intervenciones clínicas eficaces para el TCA deberían estar en la vanguardia de los esfuerzos en curso.
AGRADECIMIENTOS
Los autores reciben subvenciones de los NIH (R01AA026364, R01DA037974, P50AA012870, R21CA252916, U54AA027989 y RM1HG011558) y del Departamento de Asuntos de Veteranos (1I01CX001849). HZ también recibió el apoyo de una beca NARSAD para jóvenes investigadores (nº 27835) de la Brain & Behavior Research Foundation.
Declaración de conflicto de intereses
JG es titular de la patente estadounidense 10.900.082 titulada «Dosificación de agonistas opioides guiada por el genotipo», emitida el 26 de enero de 2021. JG es remunerado por su trabajo editorial en la revista Complex Psychiatry.


