INTRODUCCIÓN
El cáncer de mama ocupa el primer lugar desde hace más de una década como la primera causa de muerte por neoplasias malignas en las mujeres mexicanas, impactando principalmente grupos de edad entre la cuarta y sexta década de la vida y afectando a todos los niveles socioeconómicos (Grajales Pérez, et al., 2014). Las tasas de mortalidad por esta enfermedad neoplásica en la República Mexicana muestran un importante incremento en las últimas cinco décadas, ya que entre 1955 y 1960, la tasa era alrededor de dos a cuatro muertes por 100.000 mujeres, elevándose de manera constante en las mujeres adultas de todos los grupos de edad, pero con mayor impacto a partir de los 30 años de edad hasta alcanzar una cifra cercana a 9 por 100,000 habitantes para la mitad de la década de 1990 y se ha mantenido más o menos estable desde entonces. Las estimaciones a corto plazo orientan a que seguirá con la misma tendencia hacia el alza en el número de casos (Knaul, et al., 2009).
A pesar de intensas investigaciones epidemiológicas, la evidencia señala que el cáncer de mama es una enfermedad multifactorial, donde sobresalen en la población mexicana la obesidad, la inactividad física y la historia familiar como los factores de riesgo más comunes. Por otro lado, el embarazo a temprana edad y la lactancia por lo menos durante seis meses, tienen un efecto protector (Knaul, et al., 2009).
En la República Mexicana existen diferencias regionales importantes, con una mayor frecuencia de cáncer de mama en los estados del norte y centro, donde las mujeres gozan de un estado socioeconómico y cultural más elevado, mientras que en los estados donde predomina la población indígena y de menor nivel socioeconómico la frecuencia es más baja (Rodríguez Cuevas y Capurso García, 2006; Sifuentes-Álvarez, et al., 2015). Esta evidencia es compatible con la epidemiología del cáncer de mama a nivel mundial, donde afecta con mayor importancia a los países desarrollados y presenta tasas de incidencia y prevalencia mucho menores en poblaciones del tercer mundo. Más que un componente socioeconómico o ambiental, diversos autores han mencionado la relación con la genética propia de las diversas poblaciones humanas, con especial atención a los diferentes haplogrupos mitocondriales.
Se ha sugerido que las alteraciones del ADN mitocondrial juegan un papel importante en la carcinogénesis, donde la región control llamada D-loop de la secuencia ADN mitocondrial (ADNmt) contiene secuencias esenciales para la transcripción y replicación, por lo que estudios previos sugieren que polimorfismos en estas regiones no codificantes del área control pueden desempeñar un importante papel en la patogénesis del cáncer de mama (Ye, et al., 2010; Maggrah, et al., 2013). Estos cambios en la secuencia pueden asociarse con el fenotipo particular y servir como marcadores para el desarrollo de neoplasias malignas (Singh y Kulawiec, 2009).
Otros trabajos se han enfocado en polimorfismos asociados a regiones relacionadas a la síntesis de proteínas y RNA importantes para el metabolismo mitocondrial. Dentro de los más citados en la literatura, el polimorfismo G10398A asociado con síntesis de la proteína NADH-ubiquinona oxireductasa 3 (ND3) se ha identificado como un factor de riesgo para cáncer de mama en mujeres originarias de la India (Singh y Kulawiec, 2009; Francis, et al., 2013), chinas (Jiang, et al., 2014) y principalmente en poblaciones afrodescendientes (Canter, et al., 2006; Mims, et al., 2006; Setiawan, et al., 2008; Kulawiec, et al., 2009). Este mismo polimorfismo también se ha asociado a síndrome metabólico y trastornos mentales en otras poblaciones de origen asiático (Kishida, et al., 2009; Juo, et al., 2010).
Aunque en grupos humanos americanos caucásicos descendientes de europeos, la sustitución A10398G confiere un mayor riesgo de cáncer de mama, se han identificado otros con mayor significancia estadística, como el polimorfismo T16519C localizado en la región control (Bai, et al., 2007; Weigl, et al., 2013).
A pesar de estos prometedores resultados, y que la mayoría de estos polimorfismos del ADNmt tienen funcionalmente consecuencias, las asociaciones con polimorfismos específicos y el riesgo de cáncer han estado sujetas a intensos debates. Varios estudios que implican la asociación de polimorfismos específicos con riesgo de cáncer han sido minuciosamente analizados debido a un diseño experimental erróneo, interpretación y datos de baja calidad (Chatterjee, et al., 2011).
Muchas de estas variantes del ADNmt podrían no ser concluyentes debido a artefactos relacionados con errores de genotipado o un diseño experimental inadecuado (Salas, et al., 2014). Estudios previos han sido realizados mediante el análisis de los datos publicados en los estudios de asociación de casos y controles de cáncer de mama con un enfoque basado en la filogenética han empleado tanto secuencias completas de genoma como secuencias parciales que corresponden principalmente a segmentos de la región de control encontrando inconsistencias y contradicciones para el polimorfismo no sinónimo A10398G en cáncer de mama, principalmente al seleccionar grupos de control, así como en el empleo de estadística inadecuada (Salas, et al., 2014) y a errores en la nomenclatura (Mao, et al., 2013). Sin embargo, debido a la utilidad potencial como herramienta de diagnóstico, el estudio ADN mitocondrial y su relación con cáncer debe seguir siendo un foco importante de investigación de biomarcadores oncológicos en espera del diseño adecuado de estudios, la estratificación de la población y la replicación independiente de los resultados.
En este trabajo proponemos realizar el haplotipado reutilizando secuencias actualmente disponibles en bases de datos públicas y explorar la prevalencia de polimorfismos previamente asociados como marcadores de riesgo y protectores para cáncer de mama en población mestiza de origen mexicano, utilizando una población control con baja prevalencia para la enfermedad como referencia.
MATERIALES Y MÉTODOS
Población de estudio
Se realizó la búsqueda de secuencias completas de cromosomas mitocondriales (16,569 pares de bases) en la base de datos Nucleotide de Genebank (National Center for Biotechnology Information) (https://www.ncbi.nlm.nih.gov/nucleotide), sin tomar en consideración fragmentos menores a 15,400 pares de bases, debido a que la mayor parte de trabajos sobre el tema se amplifica la región control con tamaños menores a 1,000 pares de bases para estudios epidemiológicos, de correlación clínica, antropológicos poblacionales y evolutivos filogeográficos. Esta estrategia de exploración inicial tiene como objetivo obtener solo información parcial limitada que permitirá evaluar la mejor estrategia de búsqueda para más adelante explorar estas secuencias mitocondriales de la región control, las cuales son mucho más abundantes en la base de datos.
La estrategia de búsqueda se llevó a cabo empleando las palabras clave Homo sapiens, mitochondrion, Breast Cancer, Mexico y Mexican y booleanos y filtros para seleccionar los resultados, excluyendo el término “restos humanos antiguos” (ancient human remains) para facilitar la búsqueda en la base de datos.
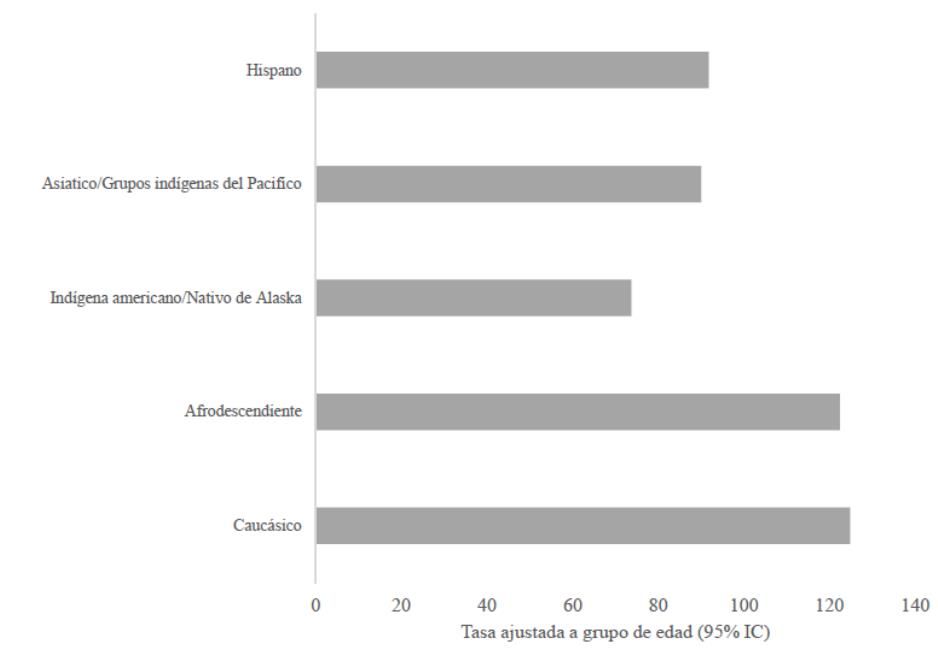
Como grupo control se empleó una población humana con baja incidencia de cáncer de mama (Figura 1), seleccionado a grupos indígenas esquimales que habitan el norte de Canadá y Alaska y empleando como palabras claves para la búsqueda Eskimo e Inuit.
Ya identificadas las secuencias en la base de datos, se procedió explorar los metadatos acompañantes de cada una de ellas para validar el lugar de origen y eliminar las que no correspondían o no especificaban el grupo étnico del individuo de nuestro interés. De las secuencias seleccionadas, se obtuvo la referencia de la cita del trabajo para evaluar el fin del muestreo y sus objetivos en el diseño experimental de cada trabajo en particular.
Una vez realizado esto, empleando el número de secuencia de Genebank se obtuvieron las secuencias completas en archivos con formato FASTA y se llevó a cabo la haplotificación y contabilización de polimorfismos mediante la aplicación MITOMASTER disponible de forma gratuita en el servidor https://www.mitomap.org/foswiki/bin/view/MITOMASTER, construyendo una base de datos en una hoja de cálculo .xlsx (Excel Microsoft) para la tabulación y análisis estadístico de las frecuencias. Los criterios para clasificación de los diferentes haplogrupos pueden encontrarse en la base de datos Phylotree (http://www.phylotree.org/) (van Oven y Kayser, 2009). Una vez realizado el haplotipado y tener identificadas las variantes en las secuencias, se construyó una base de datos para cuantificar los haplogrupos, haplotipos y subclados principales comparando las poblaciones con cáncer de mama y descendencia mexicana con la población control de origen esquimal.
Para el análisis de la estructura de las poblaciones, se construyó un solo archivo con todas las secuencias en formato FASTA que fue usado como insumo para el alineamiento de las secuencias mediante el programa CLUSTAL X (http://www.clustal.org/clustal2/). Una vez alineadas, empleando la paquetería DnaSP (http://www.ub.edu/dnasp/) se estratificó clasificando en dos poblaciones (mexicodescendientes y esquimales) con sus respectivos haplogrupos cada uno. A partir de esta segmentación, se estimó el número de loci polimórficos, el número de diferencias fijas, las mutaciones polimórficas y monomórficas entre las poblaciones, los polimorfismos compartidos y el número promedio de diferentes nucleótidos entre poblaciones. También se calcularon por el método de Jukes y Cantor la diversidad nucleotídica (Pi) entre haplogrupos, el número promedio de sustituciones de nucleótidos por sitio entre poblaciones (Dxy) y número neto de sustituciones de nucleótidos por posición entre poblaciones (Da). Se tomó en consideración la significancia estadística calculada empleando el número total de polimorfismos en las secuencias con P < 0.05. Finalmente se utilizó el programa Arlequín versión 3.5.2.2 (http://cmpg.unibe.ch/software/arlequin35/Arlequin35.html) para estimar el flujo génico entre haplogrupos mediante análisis poblacional de distancias por cálculo de FST de Wrigth considerando también la significancia estadística de las diferencias con P < 0.05.
Finalmente, se llevó a cabo la búsqueda de polimorfismos definidos previamente en la literatura asociados a mayor riesgo a cáncer de mama (T16519C, A10398G y G9055A) y los polimorfismos relacionados como protectores para desarrollo de la enfermedad (T3197C y G13708A). Para evaluar su correlación se comparó el grupo de cáncer de mama con las poblaciones de origen mexicano y esquimal empleando la prueba de X2 de Pearson .
RESULTADOS
Se identificaron 52 secuencias de ADNmt completo de pacientes con diagnóstico confirmado de cáncer de mama. La mayor parte de la información de secuencias proviene de los trabajos de Wang y cols. (Wang, et al., 2007) y Fendt y cols. (Fendt, et al., 2011), y una proporción de secuencias del reporte de Gasparre y cols. (Gasparre, et al., 2007).
Además, se obtuvieron 283 secuencias de población con descendencia mexicana de las cuales solo 250 secuencias fueron seleccionadas de acuerdo con los criterios mencionados previamente. La mayor parte proceden del estudio de Kumar y cols. (Kumar S., Bellis C., Zlojutro M., Melton PE., Blangero J., Curran JE., 2011) y en menor proporción secuencias depositadas en Genebank por Bedford y cols. (Bedford, et al., 2013), Greenspan B. de la empresa Family Tree DNA – Genealogy by Genetics, Ltd., en Houston, Texas en diciembre de 2015 y Secher, et al. de la Universidad de La Laguna, Sta. Cruz de Tenerife, España en mayo de 2012 sin estar asociadas a alguna publicación científica en la base de datos.
Así mismo, se obtuvieron 49 secuencias de individuos de poblaciones esquimales reportadas previamente en los estudios de Derbeneva y cols. (Derbeneva, et al., 2002), Tamm y cols. (Tamm, et al., 2007), Volodko y cols. (Volodko, et al., 2008), Gilbert y cols. (Gilbert, et al., 2008) y Dryomov y cols. (Dryomov, et al., 2015).
Los resultados del haplotipado en la población con cáncer de mama demostraron diferentes clados y subclados distribuidos de manera general en 15 haplogrupos (Tabla 1). El grupo más frecuente que domina la población con cáncer de mama es el haplogrupo H con 23 individuos que representan cerca de la mitad de la población, seguido del haplogrupo B en 7 individuos representando el 13.5% de la población y los haplogrupos C, N y J, con 3 individuos cada uno, que representan en su conjunto 11.4% de la población. Los haplogrupos A, B y C, usualmente asociados a poblaciones americanas se encuentran en 12 individuos del grupo con cáncer de mama que en su conjunto representan casi una cuarta parte de la población (23.1%) (Tabla 1).
Tabla 1. Resumen comparativo de distribución de haplogrupos mitocondriales en 52 mujeres con diagnóstico de cáncer de mama.
|
Haplogrupo |
Haplotipo |
Subclado |
Número de individuos |
% |
| A | A13 | A13 | 1 | 3.9 |
| A14 | A14 | 1 | ||
| B | B4b | B4b1a2 | 3 | 13.5 |
| B4c | B4c1b2a1 | 3 | ||
| B5a | B5a2a1a | 1 | ||
| C | C7a | C7a2a | 3 | 1.9 |
| D | D4j | D4j3a | 1 | 1.9 |
| F | F1a | F1a1’4 | 1 | 3.8 |
| F1a | F1a1a1 | 1 | ||
| H | H | H | 1 | 44.2 |
| H1a | H1ay | 1 | ||
| H1e | H1e5a | 1 | ||
| H1e | H1e1 | 1 | ||
| H1g | H1g1 | 1 | ||
| H1h | H1h1 | 1 | ||
| H2a | H2a | 1 | ||
| H3q | H3q1 | 1 | ||
| H3r | H3r1 | 1 | ||
| H3u | H3u1 | 1 | ||
| H5a | H5a4a1 | 1 | ||
| H5b | H5b | 1 | ||
| H5r | H5r | 1 | ||
| H7a | H7a1a | 1 | ||
| H11a | H11a5 | 1 | ||
| H13a | H13a2b1 | 1 | ||
| H44a | H44a | 1 | ||
| H65a | H65a | 1 | ||
| HV1a | HV1a2 | 1 | ||
| HV1a | HV1a’b’c | 1 | ||
| HV11a | HV11a | 1 | ||
| HV22 | HV22 | 1 | ||
| HV4a | HV4a1 | 1 | ||
| J | J1c | J1c2 | 1 | 5.8 |
| J1c | J1c2c2a | 1 | ||
| J1c | J1c11a | 1 | ||
| K | K1a | K1a1b1c | 1 | 1.9 |
| M | M35b | M35b2 | 1 | 1.9 |
| N | N1a | N1a3a3 | 1 | 5.8 |
| N1a1a1a1 | 2 | |||
| R | R0a | R0a1a3 | 1 | 3.8 |
| R9b | R9b1b | 1 | ||
| T | T2c | T2c1a2 | 1 | 1.9 |
| U | U5a | U5a2c2 | 1 | 1.9 |
| V | V2 | V2 | 1 | 1.9 |
| X | X2n | X2n | 1 | 1.9 |
| Total | 52 | 100 |
Por lo que respecta a la población mestiza mexicana, los resultados generales del haplotipado se muestran en la tabla 2. El haplogrupo A fue el que se encontró con mayor número y con mayor diversidad de subclados (37 en total), siendo las más comunes los haplotipos A2 y A2a con 20 y 11 individuos que representan el 22 y 12% respectivamente dentro de su grupo; siendo el más polimorfo el haplogrupo A2a con 8 diferentes subclados (tabla 3).
Tabla 2. Resumen de principales haplogrupos encontrados en población mexicana contemporánea.
| Haplogrupo | Frecuencia | % |
| A | 90 | 36.0 |
| B | 65 | 26.0 |
| C | 68 | 27.2 |
| D | 19 | 7.6 |
| I | 1 | 0.4 |
| L | 1 | 0.4 |
| T | 4 | 1.6 |
| U | 2 | 0.8 |
| Total | 250 | 100 |
Tabla 3. Distribución de haplogrupo A en individuos de población mexicana contemporánea. Este grupo fue el más numeroso, definiéndose 37 subclados exclusivamente limitados al haplogrupo A2.
|
Haplogrupo |
Subclado |
Individuos |
% |
% grupal |
| A2 | A2 | 8 | 8.9 | 22.2 |
| A2+64 | 9 | 10.0 | ||
| A2+64+@153 | 2 | 2.2 | ||
| A2+64+@16111 | 1 | 1.1 | ||
| A2a | A2a4 | 1 | 1.1 | 12.2 |
| A2ae | 1 | 1.1 | ||
| A2af1b2 | 2 | 2.2 | ||
| A2ai | 2 | 2.2 | ||
| A2aj | 2 | 2.2 | ||
| A2ao | 1 | 1.1 | ||
| A2ao1 | 1 | 1.1 | ||
| A2ap | 1 | 1.1 | ||
| A2c | A2c | 1 | 1.1 | |
| A2d | A2d | 5 | 5.6 | |
| A2f | A2f2 | 2 | 2.2 | |
| A2f3 | 2 | 2.2 | ||
| A2g | A2g | 4 | 4.4 | |
| A2g1 | 2 | 2.2 | ||
| A2h | A2h1 | 1 | 1.1 | |
| A2j | A2j | 2 | 2.2 | |
| A2j1 | 1 | 1.1 | ||
| A2l | 4 | 4 | 65.6 | |
| A2m | 3 | 3 | ||
| A2o | 2 | 2 | ||
| A2p | A2p | 3 | 3.3 | |
| A2p1 | 4 | 4.4 | ||
| A2q | A2q1 | 1 | 1.1 | |
| A2r | A2r | 1 | 1.1 | |
| A2r1 | 1 | 1.1 | ||
| A2s | 1 | 1 | ||
| A2t | 4 | 4 | ||
| A2u | A2u1 | 5 | 5.6 | |
| Au2 | 2 | 2.2 | ||
| A2v | A2v1a | 1 | 1.1 | |
| A2v1+152 | 3 | 3.3 | ||
| A2w | 1 | 1 | ||
| A2x | 3 | 3 | ||
| Total | 90 | 100 | ||
El haplogrupo B, conformado por 65 individuos que representan el 26% del total de la población analizada, fue el siguiente grupo en diversidad con 30 subclados diferentes, aunque la mitad de su población se encuentra distribuida en los haplotipos B2a y B2c (Tabla 4).
Tabla 4. Distribución de haplogrupo B en población mexicana contemporánea. Este grupo registró la mayor diversidad con 30 subclados exclusivamente limitados al haplogrupo B2.
| Haplogrupo |
Subclado |
Individuos |
% |
% grupal | |
| B2 | B2 | 5 | 7.7 | 13.8 | |
| B2+16278 | 4 | 6.2 | |||
| B2a | B2a | 1 | 1.5 | 23.1 | |
| B2a1 | 1 | 1.5 | |||
| B2a1a1 | 2 | 3.1 | |||
| B2a1b | 1 | 1.5 | |||
| B2a2 | 1 | 1.5 | |||
| B2a3 | 5 | 7.7 | |||
| B2a4a | 1 | 1.5 | |||
| B2a4a1 | 3 | 4.6 | |||
| B2b | B2b4 | 1 | 1.5 | 1.5 | |
| B2c | B2c1 | 2 | 3.1 | 27.7 | |
| B2c1a | 3 | 4.6 | |||
| B2c1b | 1 | 1.5 | |||
| B2c1c | 1 | 1.5 | |||
| B2c2 | 2 | 3.1 | |||
| B2c2a | 6 | 9.2 | |||
| B2c2b | 3 | 4.6 | |||
| B2f | 2 | 1 | 33.8 | ||
| B2g | B2g1 | 2 | 3.1 | ||
| B2k | 4 | 2 | |||
| B2m | 2 | 1 | |||
| B2n | 2 | 1 | |||
| B2p | 2 | 5 | |||
| B2q | 3 | 4 | |||
| B2s | B2s1 | 1 | 1.5 | ||
| B2u | 2 | 1 | |||
| B2v | 3 | 2 | |||
| B2w | 1 | 1 | |||
| B2x | 2 | 1 | |||
| Total | 85 | 100 | |||
Por lo que respecta al haplogrupo C se encontró en 68 individuos representando el 27% de la población estudiada (Tabla 5) y estructurado en 22 subclados exclusivamente limitados al haplotipo C1 y con la mayor parte distribuidos en el subclado C1b (Tabla 8). El haplogrupo D estaba estructurado 19 individuos dispersos en 14 subclados, constituyeron el 7.6% de nuestro universo de estudio (Tabla 2), sobresaliendo que este fue el único grupo en el que su población se encontró distribuida en dos clados, donde la mayor parte en el subclado D1 y una pequeña proporción en el subclado D4 (Tabla 6).
Tabla 5. Distribución de haplogrupo C en población mexicana contemporánea, estructurado en 22 subclados exclusivamente limitados al haplogrupo C1 y con la mayor parte distribuidos en el subclado C1b.
| Haplogrupo | Subclado | Individuos | % |
% grupal |
|
| C | 15 | 22.4 | 22.1 | ||
| C1b | C1b1 | 3 | 4.5 | 42.6 | |
| C1b5a | 1 | 1.5 | |||
| C1b5b | 1 | 1.5 | |||
| C1b7 | 3 | 4.5 | |||
| C1b7a | 3 | 4.5 | |||
| C1b8a | 3 | 4.5 | |||
| C1b9 | 6 | 9.0 | |||
| C1b10 | 5 | 7.5 | |||
| C1b12 | 2 | 3.0 | |||
| C1b14 | 2 | 3.0 | |||
| C1c | C1c | 1 | 1.5 | 23.5 | |
| Cic1b | 1 | 1.5 | |||
| C1c2 | 7 | 10.3 | |||
| C1c4 | 2 | 2.9 | |||
| C1c5 | 2 | 2.9 | |||
| C1c6 | 1 | 1.5 | |||
| C1c7 | 2 | 2.9 | |||
| C1d | C1d1 | 1 | 1.5 | 11.8 | |
| C1d1a | 1 | 1.5 | |||
| C1d1C | 1 | 1.5 | |||
| C1d1c1 | 5 | 7.4 | |||
| Total | 68 | 100 | |||
Tabla 6. Distribución de haplogrupo D en población mexicana contemporánea, estructurado en 14 subclados. Este fue el único grupo en el que su población se encontró distribuida en dos subclados, donde la mayor parte en el subclado D1 y una pequeña proporción en el subclado D4.
| Haplogrupo | Subclado | Individuos | % | % grupal |
| D1 | 3 | 15.8 | 15.8 | |
| D1c | 2 | 10.5 | 10.5 | |
| D1d | D1d1 | 1 | 5.3 | 10.5 |
| D1d2 | 1 | 5.3 | ||
| D1f | D1f3 | 1 | 5.3 | 5.3 |
| D1h | D1h1 | 2 | 10.5 | 15.8 |
| D1h1 | 1 | 5.3 | ||
| D1i | D1i | 1 | 5.3 | 15.8 |
| D1i1 | 1 | 5.3 | ||
| D1i2 | 1 | 5.3 | ||
| D1k | 1 | 5.3 | 26.3
|
|
| D1m | 1 | 5.3 | ||
| D4e | 2 | 10.5 | ||
| D4h | 1 | 5.3 | ||
| Total | 19 | 100 | ||
Se encontraron subclados exóticos no comunes en la población mestiza como los haplotipos I5a, T2e y U6a, representando en conjunto menos de 2.0 del total (Tabla 7). Haplogrupos L asociados a población afrodescendiente solo se encontraron en dos individuos contribuyendo solo con el 0.8%, el cual representó el haplogrupo menos representado en el universo estudiado tomando en cuenta la gran proporción de población afrodescendiente que actualmente radica México. Marcadores asociados a poblaciones europeas como el haplogrupo H, no fueron encontrados en el análisis de las poblaciones mexicodescendientes (Tabla 2).
Tabla 7. Haplogrupos exóticos en población mexicana contemporánea. El haplogrupo T2e corresponde a origen euroasiático asociado a judíos sefarditas. El haplogrupo L asociado a poblaciones afrodescendientes está prácticamente ausente y subrepresentado en la muestra de población estudiada.
| Haplogrupo | Subclado | Frecuencia | % |
| I5a | I5a2 | 1 | 12.5 |
| L2d | L2d+16129 | 1 | 12.5 |
| T2e | T2e1a1a | 4 | 50.0 |
| U6a | U6a7a1b | 1 | 12.5 |
| U5b | U5b1g | 1 | 12.5 |
| 8 | 100.0 |
Finalmente, por lo que respecta al haplotipado de la población control esquimal, se encontró distribuida en tres haplogrupos solamente: A B y D, donde tres cuartas partes de sus integrantes se encontraron distribuidas dentro del haplogrupo A (Tabla 8). El haplogrupo A2 se encontró con mayor número y diversidad de subclados, 4 en total, dominado por el haplogrupo A2a con 17 individuos distribuidos en tres haplotipos A2a2 y A2a3 y A2b1 que en su conjunto representan el 35% del grupo respectivamente.
El haplogrupo D, el siguiente en importancia, conformado por 16 individuos que representan una tercera parte de la población esquimal total, y por lo que respecta al haplogrupo C se encontró subrepresentado solo con un individuo que representó solo menos del 25 del grupo. No se encontraron individuos con haplogrupo B en la población esquimal (Tabla 8).
Tabla 8. Haplogrupos en población esquimal originaria de Norte América. Los haplogrupos A2 y D2 son los más comunes y con una distribución muy similar a otras poblaciones indígenas y mestizas originarias del continente americano.
| Haplogrupo | Subclado | Frecuencia | % |
| A2 | A2a | 11 | 22.4 |
| A2a2 | 3 | 6.1 | |
| A2a3 | 3 | 6.1 | |
| A2b1 | 15 | 30.6 | |
| C4 | C4b2a | 1 | 2.0 |
| D | D2a1 | 1 | 2.0 |
| D2a1b | 8 | 16.3 | |
| D2a2 | 5 | 10.2 | |
| D4b1a2a1 | 2 | 4.1 | |
| 49 | 100.0 |
Por lo que respecta a los polimorfismos identificados en las tres poblaciones estudiadas se encuentran en todas las secuencias analizadas, en promedio entre 26 y 39 polimorfismos dependiendo el grupo analizado (Tabla 9). En el grupo mexicodescendiente se encontró el mayor número de polimorfismos en secuencias de genomas mitocondriales (61) mientras que en el grupo de cáncer de mama se encontraron con menor frecuencia (11).
Tabla 9. Resumen de polimorfismos identificados en secuencias de ADNmt en tres diferentes en dos diferentes grupos poblacionales y mujeres con cáncer de mama.
|
Población |
Individuos |
Número máximo de polimorfismos por secuencia |
Número mínimo de polimorfismos por secuencia |
Promedio de polimorfismos por secuencia |
| Mexico-descendientes | 250 | 61 | 28 | 39 |
| Esquimales | 49 | 47 | 30 | 35 |
| Cancer de mama | 52 | 47 | 11 | 26 |
El análisis de la estructura de las poblaciones estudiadas nos muestra que el número de loci polimórficos varía entre 364 y 116, encontrándose solo 6 polimorfismos fijos tanto en la población mexicodescendiente como en la población esquimal en el Haplogrupo C, siendo más comunes las mutaciones polimórficas en mexicodescendientes que en esquimales (Tabla 10). Por lo que respecta a polimorfismos compartidos, se encontraron 7 y 8 en los haplogrupos A y D respectivamente, mientras que estuvieron ausentes en el haplogrupo C y el número promedio de diferentes nucleótidos entre poblaciones disminuye entre más numeroso es el haplogrupo analizado (Tabla 10). La prueba de neutralidad de Tajima (D) muestra un coeficiente menor a 0 con significancia estadística sólo en los haplogrupos A y C de ambas poblaciones (Tabla 10), lo que se interpreta como la presencia de alelos raros poco comunes en altas frecuencias, compatible con la biología mitocondrial, con una elevada frecuencia de replicación, con mecanismos de reparación ineficientes y poca protección a agentes mutagénicos, lo que permite una alta proporción de polimorfismos. Por otro lado, el análisis de aislamiento por distancia que se muestra en la tabla 11, hace evidente un valor de Fst al comparar los haplogrupos A de las poblaciones de origen mexicano y esquimal. Presenta valores entre 0.15 a 0.25, lo que se interpreta como una diferenciación grande, mientras que el resto de los haplogrupos analizados presentan valores mayores a 0.25, lo que se interpreta como una diferenciación muy grande. Esta evidencia sugiere que existe una gran diferencia entre los haplogrupos estudiados debido a la fijación de alelos alternos en las diferentes poblaciones.
Tabla 10. Análisis comparativo entre diferentes haplogrupos de genoma mitocondrial entre poblaciones de origen mexicano y esquimales. Para la matriz comparativa de diversidad nucleotídica Pi entre haplogrupos se realizó mediante el método de Jukes & Cantor. Para la estimación de los estadísticos se utilizaron los programas DnaSP (http://www.ub.edu/dnasp/) y Arlequin (http://cmpg.unibe.ch/software/arlequin35/).
| Tipo de análisis comparativo entre poblaciones | A | C | D |
| Número de loci polimórficos | 324 | 216 | 116 |
| Número de diferencias fijas | 0 | 6 | 0 |
| Mutaciones polimórficas en mexicodescendientes, pero monomórficas en esquimales | 303 | 210 | 77 |
| Mutaciones polimórficas en esquimales, pero monomórficas en mexicodescendientes | 14 | 0 | 31 |
| Polimorfismos compartidos | 7 | 0 | 8 |
| Número promedio de diferentes nucleótidos entre poblaciones | 9.999 | 15.888 | 18.531 |
| Número promedio de sustitución de nucleótidos por sitio entre poblaciones (Dxy) | 0.00060 | 0.00094 | 0.00112 |
| Numero neto de sustitución de nucleótidos por posición entre poblaciones(Da) | 0.00010 | 0.00053 | 0.0005 |
| Prueba de neutralidad de Tajima (D) | -2.45389* | -2.10103** | -1.52697 |
Para el cálculo de Dxy y Da se empleó el método de Jukes & Cantor.
*Significancia estadística P < 0.001 calculada empleando el número total de polimorfismos en las secuencias.
**Significancia estadística P < 0.05 calculada empleando el número total de polimorfismos en las secuencias.
| Pi Población mexicodescendiente | ||||||
| Haplogrupos | A | B | C | D | ||
| Pi Población esquimal | ||||||
| A | 0.00067 | 0.00142 | 0.00158 | 0.00126 | ||
| C | 0.00081 | 0.00086 | 0.00083 | 0.00083 | ||
| D | 0.00124 | 0.00148 | 0.00124 | 0.00071 | ||
Tabla 11. Estimación de flujo génico entre haplogrupos de genoma mitocondrial mediante análisis poblacional de distancias por cálculo de FST de Wrigth. Los subíndices se refieren a las poblaciones de origen mexidescendiente (m) y esquimal (e). La estimación se realizó empleando el programa Arlequin (http://cmpg.unibe.ch/software/arlequin35/).
| Mexicodescendientes | Esquimales | ||||||||
| Am | Bm | Cm | Dm | Ae | Ce | De | |||
| Mexico-descendientes | Am | 0.00000 | |||||||
| Bm | 0.71058* | 0.00000 | |||||||
| Cm | 0.71540* | 0.76328* | 0.00000 | ||||||
| Dm | 0.67984* | 0.73531* | 0.58452* | 0.00000 | |||||
| Esquimales | Ae | 0.13851* | 0.76670* | 0.76499* | 0.79944* | 0.00000 | |||
| Ce | 0.74253* | 0.77064* | 0.39013* | 0.66676* | 0.90153* | 0.00000 | |||
| De | 0.71215* | 0.76918* | 0.65766* | 0.41021* | 0.84208* | 0.76179* | 0.00000 | ||
Después de la estimación de Dxy y Da, es posible identificar los puntos físicos donde existe mayor diversidad y divergencia entre las poblaciones (Figura 2).
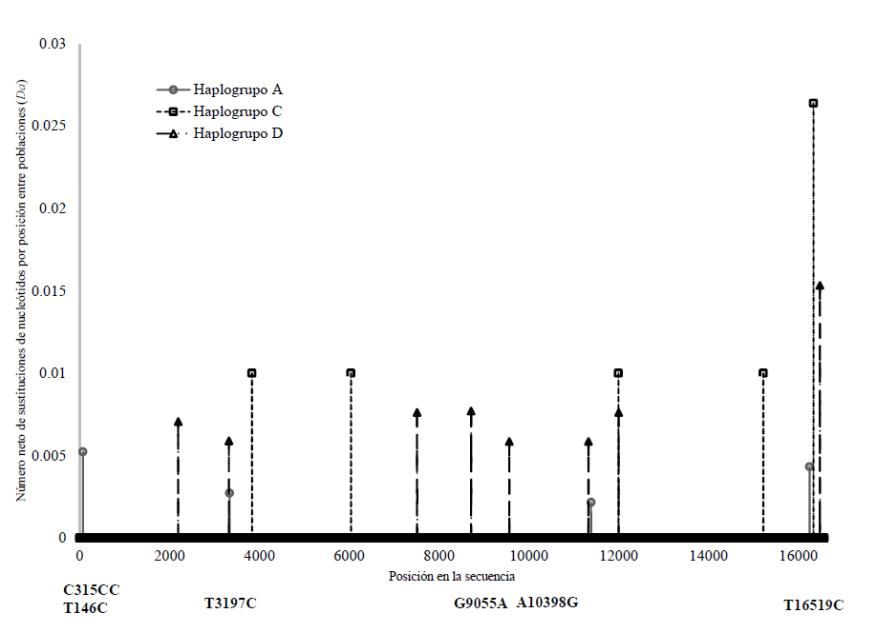
Por lo que respecta a polimorfismos asociados a mayor riesgo para el desarrollo de cáncer de mama se identificaron tres descritos previamente en la literatura (T16519C, A10398G y G9055A) (Bai, et al., 2007; Covarrubias, et al., 2008). Además se encontró una probable relación con el polimorfismo C315CC, mostrando sus proporciones y razones en la tabla 12. De igual manera, se localizaron dos polimorfismos previamente definidos como protectores (T3197C y G13708A) y una probable asociación identificada en nuestro análisis con T146C. Al explorar la correlación entre estos polimorfismos presentes mediante el coeficiente de correlación de Pearson se demostró asociación directa con significancia estadística entre el grupo con cáncer de mama y las poblaciones de origen mexicano (Tabla 12).
Tabla 12. Análisis de polimorfismos en secuencias de ADNmt asociados con mayor riesgo y protección contra cáncer de mama. Para estimar la significancia estadística entre las variables se realizó la prueba de X2 para evaluar cada uno de los polimorfismos en cada una de las poblaciones. Para estimar la relación entre las variables se evaluaron los polimorfismos de la población de referencia con cáncer de mama y las poblaciones estudiadas de origen mexicano y esquimal estimando el coeficiente de correlación de Pearson.
* p<0.05
** Identificados en este estudio.
|
Polimorfismos |
Cancer de mama (%) |
Mexicodescendientes (%) |
Esquimales (%) |
Totales |
| Número de individuos en la población analizada | 52 (100) | 250 (100) | 48 (100) | 350 |
| Número de polimorfismos por población | 83 | 370 | 55 | 508 |
| Mayor riesgo | ||||
| T16519C | 33 (63.5) | 112 (44.8) | 2 (4.2) | 147 |
| A10398G | 11 (21.2) | 90 (36.0) | 17 (35.4) | 118 |
| G9055A | 1 (1.9) | 3 (1.2) | 0 | 4 |
| C315CC** | 26 (50.0) | 118 (47.2) | 4 (8.3) | 2 |
| Protectores | ||||
| T3197C | 1 (1.9) | 1 (0.4) | 0 | 2 |
| G13708A | 3 (5.8) | 1 (0.4) | 0 | 4 |
| T146C** | 8 (15.4) | 45 (18) | 32 (66.7) | 85 |
| Coeficiente de correlación de Pearson | – | 0.914997713* | -0.04172622* |
DISCUSIÓN
Aparentemente no existe relación entre los diferentes haplogrupos y los polimorfismos asociados con riesgo o protección contra cáncer de mama. Sin embargo, al analizar la información podemos observar que el número de polimorfismos por secuencia son menos frecuentes en el grupo de mujeres con cáncer de mama. Este es probablemente un sesgo estadístico debido a que la mayoría de los individuos son de origen europeo, ya que cerca de la mitad de la población con cáncer de mama (44.9%) pertenecen al haplogrupo H, común en poblaciones de descendencia europea, y estas son más similares a la secuencia consenso que se utiliza como genoma de referencia que se emplea en la base de datos y en los navegadores genómicos. Esta secuencia de referencia se le conoce como revised Cambridge Reference Sequence ó rCRS y en GenBank es la secuencia NC_012920.
De los polimorfismos asociados con riesgo para desarrollar la enfermedad, sólo T16519C tuvo correlación estadística, encontrándose con mayor frecuencia en las poblaciones con cáncer de mama y descendientes de mexicanos, con 63.5 y 44.8% respectivamente, mientras que en la población esquimal su prevalencia era baja, menor al 4.2%. Por otro lado, los polimorfismos A10398G y G9055A no tuvieron asociación estadística en nuestro análisis.
Pudimos identificar un polimorfismo no descrito en la literatura asociado probablemente a mayor riesgo, C315CC encontrándose en 26 individuos (50.0%) del grupo con cáncer de mama, en 118 individuos (47.2%) del grupo mestizo de origen mexicano y solo en 4 individuos (8.3%) del grupo de origen esquimal. Se requiere mayor análisis e incrementar el número de secuencias para estratificar adecuadamente y establecer si existe correlación con mayor riesgo para el desarrollo de la neoplasia maligna. También es importante hacer notar que es probable la asociación simultánea de varios polimorfismos en una misma secuencia, por lo que requiere replantear el análisis estadístico para identificar probables correlaciones.
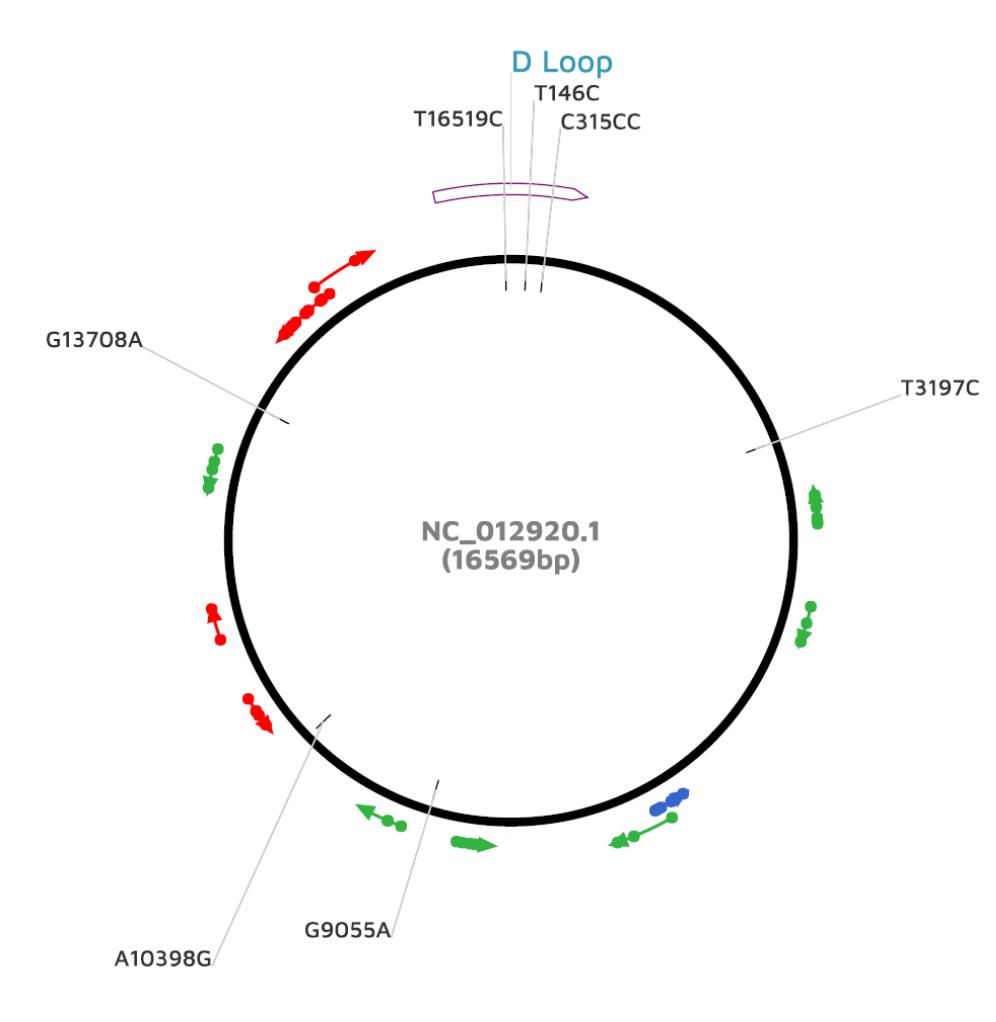
En la figura 3 se muestra el ADN mitocondrial donde se muestra la posición de la región control D Loop con respecto a los polimorfismos que de acuerdo a nuestro análisis correlacionan con mayor riesgo a desarrollar cáncer de mama: T16519C y C315CC, los cuales se encuentra en la región no codificante de la región control, mientras que A10398G y G9055A, los cuales no tuvieron correlación estadística en nuestro estudio, se encuentran asociados a pautas abiertas de lectura entre un codón de inicio (AUG) de la traducción y un codón de terminación.
Los polimorfismos protectores están ausentes en las tres poblaciones analizadas, mientras que el polimorfismo T146C se encuentra con una baja prevalencia en la población con cáncer de mama, identificado solo en 8 individuos (15.4%) y en la población mestiza en 45 individuos (18%), mientras que en la población esquimal se identificó en dos terceras partes del grupo (32 individuos que conformaron en 66.7% del total del grupo).
Este primer ejercicio es un acercamiento en el que sólo empleamos secuencias completas del cromosoma mitocondrial. En trabajos futuros podemos incluir el gran acervo que existe en Genebank de fragmentos de la región control del cromosoma mitocondrial (este tiene 69,557 secuencias registradas hasta el día 6 de enero del 2018 (https://www.mitomap.org/foswiki/bin/view/MITOMAP/GBFreqInfo). Esta aproximación nos daría una perspectiva general más cercana, consolidando mucha de la información dispersa en la base de datos y que no se ha cruzado hasta el momento. Esta misma estrategia se puede hacer con otros grupos humanos latinoamericanos con mezclas étnicas complejas.
Finalmente, una vez que se incremente el esfuerzo de muestreo para mayores coberturas de grupos humanos poco representados y mejores diseños experimentales para trabajo de campo de poblaciones representativas de diversos grupos indígenas, mestizos y afrodescendientes, tanto del pasado como contemporáneas, nos darán una mejor perspectiva al compararse con la información disponible actualmente.
CONFLICTO DE INTERESES
Los autores del trabajo manifiestan ningún conflicto de interés relacionado.

