INTRODUCCIÓN
La atresia de esófago (AE) es una de las malformaciones congénitas mayores más frecuentes, con una incidencia calculada de 2,1/10.000 recién nacidos vivos (Bermejo et al, 1996). Se define como la falta o ausencia de un fragmento de esófago, de modo que se origina un segmento superior ciego y una porción distal que queda como un cabo atrésico. Se pueden clasificar de tres modos: a) con criterio anatómico, que es la clasificación clásica de Gross (Figura 1), basada en la presencia y localización de la atresia y la fístula (Gross y Piotti, 1953); b) según si se presenta como malformación aislada (AE “no sindrómica”) o asociada a otras anomalías congénitas (AE compleja o “sindrómica”); c) en función de si forma parte o no de un síndrome genético conocido (de Jong et al, 2010). A efectos de manejo médico-quirúrgico, se utiliza habitualmente la clasificación de Gross, siendo la AE más frecuente la tipo III, que es la que presenta fístula traqueoesofágica inferior con un segmento ciego superior. A efectos de diagnóstico clínico y asesoramiento genético, es muy útil la distinción entre AE sindrómica y AE no sindrómica.
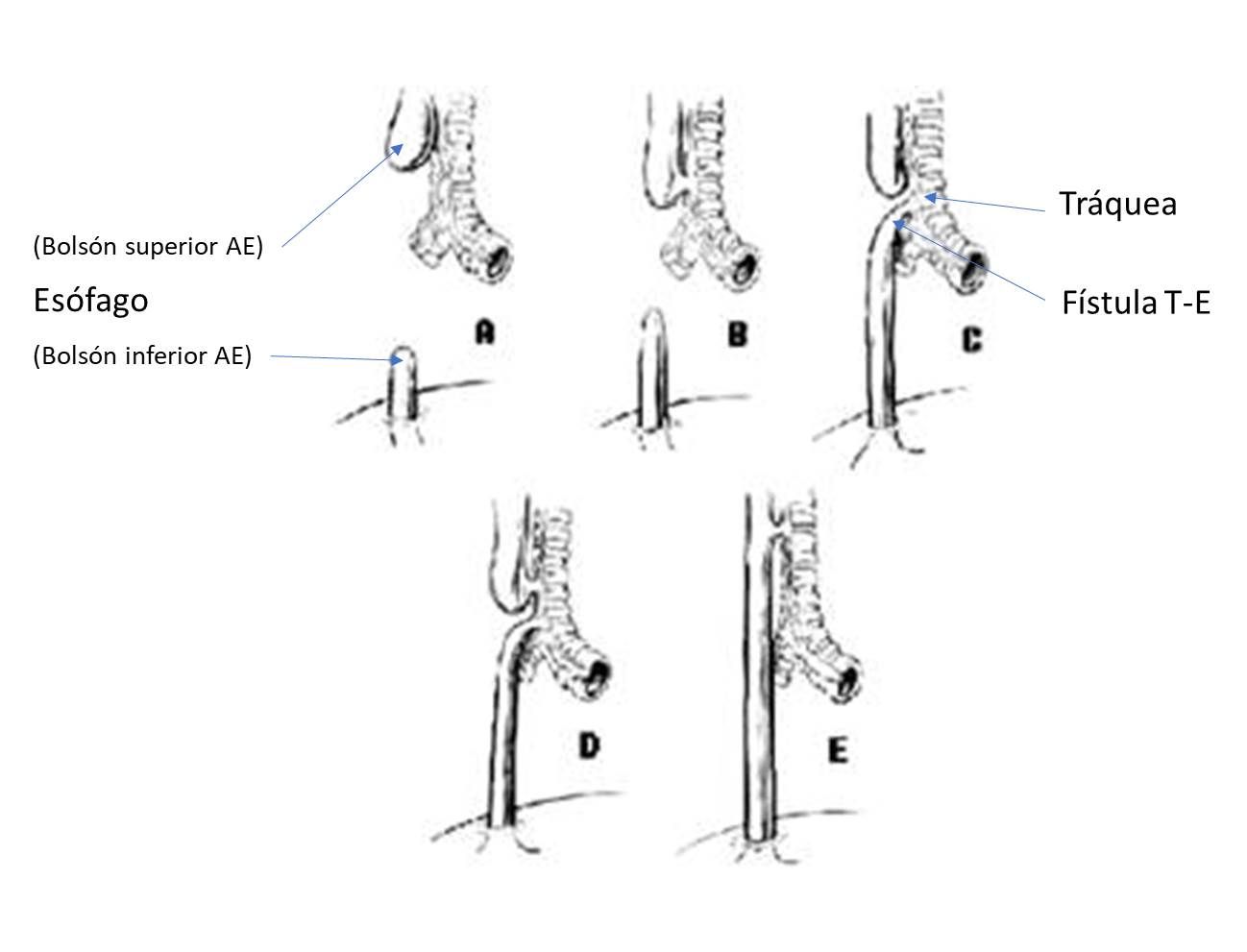
Desarrollo esofágico embrionario y Atresia de Esófago pre- y posnatal
En el embrión humano, la formación del esófago se produce al tiempo que se forma el divertículo respiratorio, o yema pulmonar, desde la pared ventral del intestino anterior, hacia la cuarta semana de desarrollo posconcepcional. Posteriormente, aparece el tabique traqueoesofágico, que separa el primordio respiratorio y el tubo digestivo. Una teoría postula que la tráquea se forma como un órgano independiente como resultado de su rápido crecimiento longitudinal (Merei et al., 1998). Por contra, otros autores defienden que la tráquea crece inicialmente unida al intestino y se convierte en una estructura separada cuando se forman los esbozos pulmonares y se produce un crecimiento en dirección craneal (Qi BQ et al., 2000). En cualquier caso, este proceso parece bastante determinado por la expresión del gen SHH (Sonic hedgehog) y otros relacionados con su cascada de señalización como FGF1 y FGF10 (Ioannides et al., 2003; Morrisey and Hogan, 2010). La diferenciación del intestino primitivo, en lo que se refiere a su zona anterior de la que derivan esófago y estómago, está controlada por diferentes genes HOS, como HOXA3 y HOXA4, y por los factores de transcripción SOX2 y NKS2.1, todos los cuales se expresan mas específicamente en intestino superior, así como también por SHH el cual se expresa a lo largo de todo el endodermo (Faure y de Santa Barbara et al., 2011). La formación del esbozo pulmonar estaría muy determinada por la sobreexpresión de NKX2.1 en la zona ventral del intestino anterior (Morrisey and Hogan et al., 2010). La separación entre ambas, la yema pulmonar y el intestino, se ha relacionado con BARX1, un factor de transcripción con fuerte expresión en el mesénquima (Woo et al., 2011). Es interesante señalar, que mutaciones en alguno de estos genes causan síndromes en los que la AE forma parte de su espectro fenotípico, un ejemplo de esto sería el síndrome AEG, producido por mutaciones en gen SOX2 y la displasia alveolocapilar asociada a FOXF1 (Tabla 1).
Tabla 1. Síndromes relacionados con AE (Modificado de Brosens et al, 2014 ).
|
Síndrome (OMIM) |
Otras malformaciones características |
Gen/es asociados |
Frecuencia de AE |
| AEG (# 206900) |
Anoftalmia/microftalmia Anomalías en genitales |
SOX2 | 100% |
|
Feingold (# 164280)
|
Microcefalia
Sindactilia 3º-4º dedos pies Hipoplasia distal extremidades |
MYCN | 25-40% |
|
CHARGE (# 214800) |
Atresia de coanas Coloboma iris/fundus Malformación oido medio (Anomalía de Mondini) Alteraciones en nervios craneales |
CHD7, SEMA3E | 25% |
|
Fanconi (# 227650) |
Craneofacial característico
Defectos de eje radial (Hipoplasia/agenesia de radio; íd primer dedo; Íd primer metacarpiano) Malf Uro-genitales Talla baja Alteraciones hematológicas |
FANC-A,B,C,D1, E, F | 8-10% |
|
Opitz GBBB (# 300000)
|
Fisura labial/palatina
Anomalías genitales (Hipospadias) Hipertelorismo |
MID1 | 0,5-1% |
| Displasia alveolocapilar (# 265380) | Anomalía en tejido pulmonar | FOXF1 | Ocasional
(1%) |
| McKusick-Kaufman (# 236700) | Polidactilia
Malformaciones genito-urinarias |
MKKS | Ocasional
(1%) |
| Pallister Hall (# 146510) | Hamartoblastoma hipotalámico
Polidactilia; Ano imperforado |
GLI3 | Ocasional
(1%) |
| TAR (# 274000) | Agenesia de radios; Trombopenia | RBM8 | Ocasional
(1%) |
| Disostosis mandibulofacial-microcefalia
(# 610536) |
Micrognatia, Microcefalia
|
EFTUD2 | Ocasional
(1%) |
| Espectro Oculo-Auriculo-Vertebral*
(O-A-V) (# 164210) |
Microsomia hemifacial
Fisura labial/palatina Malformacion pabellones auriculares |
Sin gen relacionado | Ocasional
(1%) |
| Asociación VACTERL* | Malformaciones vertebrales
Cardiopatia congénita Malformaciones tracto urinario Malformaciones extremidades (Defectos en eje radial) |
Sin gen relacionado | 70% |
Las teorías sobre las alteraciones en la organogénesis se han basado en estudios en modelos de ratas expuestas a adriamicina en las que se producía AE y fístula traqueoesofágica (Possogel et al., 1998; Ioannides et al., 2002). Muchos estudios señalan que el fallo es primario, debido a una persistencia entre la unión del árbol pulmonar y el esbozo digestivo, más que a un defecto secundario en el crecimiento traqueal (Ioannides et al., 2003). No obstante, existe también la hipótesis de que la atresia proximal del esófago es el evento primario y establece la continuidad entre la tráquea y el esófago distal (Crisera et al., 1999).
No hay marcadores biológicos específicos de AE que permitan el diagnóstico prenatal, el cual se realiza mediante la ecografía fetal, que está basada en imágenes indirectas, fundamentalmente la presencia de polihidramnios junto a la ausencia de relleno gástrico, aunque en los casos de AE con fístula (85-90%) el estómago fetal puede observarse relleno. La tasa de detección varía entre un 10 y un 50% según los centros (Pedersen et al., 2012). La medida de alfa-feto proteína y gamma-glutamil transpeptidasa en líquido amniótico, así como la resonancia magnética maternofetal en semana 32, pueden ayudar al diagnóstico prenatal (Czerkiewicz et al., 2011).
Tras el nacimiento, la AE debe ser sospechada en recién nacidos (RN) con un exceso de salivación y atragantamiento con distrés respiratorio. El diagnóstico se confirma con la imposibilidad de pasar la sonda orogástrica hasta el estómago, y la verificación de este hecho mediante una radiografía de tórax. Estos RN requieren un manejo específico en unidades de cuidados intensivos neonatales y tratamiento quirúrgico, en general en las primeras 48 horas de vida (Zani et al., 2014). Los avances en el manejo perinatal y el desarrollo de nuevas técnicas quirúrgicas han posibilitado que la supervivencia a largo plazo de los niños afectos sea mayor del 90% (Lilja y Wester, 2008). La incidencia a largo plazo de problemas gastrointestinales, como reflujo gastroesofágico, anemia o infecciones respiratorias, oscila entre un 25 y 42% de los pacientes. En cualquier caso, el principal factor que condiciona el pronóstico son las malformaciones asociadas, y tambien las alteraciones genéticas relacionadas (de Jong et al., 2010).
Atresia de esófago aislada o no sindrómica
En torno a un 50% de los pacientes, se presentan como AE aislada (Shaw-Smith et al., 2010), y aunque se ha intentado relacionar algún caso con microdelecion en el cluster FOX, la AE como malformacion aislada, no sindrómica, no se ha podido asociar hasta ahora a ninguna causa monogénica concreta (Shaw-Smith et al., 2010). No obstante, el desarrollo de modelos animales ha posibilitado explorar algunas vías que en un futuro podrían identificar genes relacionados con esta malformación. En el ratón, mutaciones en el gen Shh y otros genes de la misma vía de señalización, como Gli2, Gli3 y Foxf1, se han relacionado con AE y fístula traqueoesofágica (Litingtung et al., 1998) y por otra parte alteraciones en esta misma vía, también en modelos de ratón, han dado lugar a un espectro de anomalías similares a la asociación VACTERL (Kim et al., 2001). Además, factores de señalización del mesénquima circundante (WNt2, Wnt2b, Fgf10 y Bmp4), así como de la notocorda (Nog, Shh) parecen regular la diferenciación del epitelio para el proceso de separación (Jacobs et al., 2012).
En cualquier caso, existen datos que sugieren la existencia de una base genética en la AE, como es la concordancia en gemelos monocigóticos (67%) frente a dicigóticos (42%) (Schultz et al., 2012), la relación con síndromes monogénicos o en pacientes con alteraciones cromosómicas (Bednarcyk et al., 2013), así como los casos descritos de AE familiar, con una incidencia aproximada de 1,7% (Choinitzki et al., 2013).
Atresia de esófago compleja o sindrómica
En aquellos casos de atresia de esófago sindrómica o compleja, es decir, asociada a otras malformaciones, las anomalías más frecuentes son las relacionadas con la asociación VACTERL (En inglés: Vertebral, Anorrectal, Cardiac, Tracheo-Esophageal, Renal and Limb) (de Jong et al, 2008). Un 25% de los nacidos con AE compleja, presentarán alguna malformación de este grupo, siendo las más habituales la cardiopatía congénita y las malformaciones vertebrales (Stoll et al., 2009; Brosens et al.,2014). En todos los casos, el diagnóstico de asociación VACTERL debe ser de exclusión, ya que existen diferentes síndromes genéticos bien conocidos con fenotipos similares (Zhang et al., 2017). Entre un 10-15% de AE presentan malformaciones no incluidas en asociación VACTERL, como son otras anomalías digestivas, craneofaciales, oculares o del sistema nervioso central (Stoll et al., 2009; de Jong et al., 2008), por lo que se recomienda que ante un paciente con AE se realice, además de un examen físico detallado, un despistaje de otras anomalías congénitas con ecocardiografía, ecografía cerebral y abdominal, radiografía vertebral y de extremidades, y estudio de fondo de ojo (Beauregard-Lacroix et al., 2017).
Atresia de esófago compleja o sindrómica, relacionada con alteraciones genéticas
Es conocido desde hace ya bastante tiempo que la AE puede aparecer asociada a ciertas aneuploidías, fundamentalmente en trisomía 21 (síndrome de Down), 18 (síndrome de Edwards) y 13 (síndrome de Patau) (Brosens et al, 2014). Los actuales estudios con cariotipo molecular, han permitido identificar algunas regiones donde aparece con mayor frecuencia AE, como son las microdeleciones 2q37, 4q35, 6q13-15 y 22q11, y microduplicaciones 3p25-pter y 5q34-qter (Felix et al., 2007) (Tabla 2).
Tabla 2. Anomalías cromosómicas más frecuentemente asociadas a AE.
| Anomalía | Fenotipo mas característico |
| Trisomía 21 | – Dismorfía facial típica.
– Hipotonía. – Cardiopatía congénita (Canal A-V común). – Atresia duodenal (Páncreas anular). |
| Trisomía 18 | – Dismorfía facial típica.
– Mano “trisómica” (dedos 2º,3º, y 4º-5º, de manos, montados unos sobre otros). – Calcáneo prominente. – Cardiopatía congénita. – Hipertonía inicial que evoluciona a hipotonía. |
| Trisomía 13 | – Craneofacial dismórfico.
– Fisura labial. – Zonas de aplasia cutis en cráneo. – Polidactilia. – Poliquistosis renal. |
| Microdeleción 22q11 | – Dismorfia facial típica.
– Fisura palatina/Incompetencia palatina. – Cardiopatía congénita (Anomalías cono-truncales). – Hipoparatiroidismo (Hipocalcemia). – Deficit linfocitos T. |
| Microdeleciones:
2q37 4q35 6q13-15 |
Fenotipo puede ser inespecífico en periodo recién nacido. |
| Microduplicaciones:
3p25-pter 5q34-qter |
Fenotipo puede ser inespecifico en periodo recién nacido. |
Microdeleciones en 16q24.1, que comprenden el clúster de factores de transcripción FOX, producen malformaciones en tejido y venas pulmonares que constituyen el síndrome conocido como Displasia alveolocapilar con mal alineamiento de venas pulmonares (ACD/MPV; OMIM: 265380). Este síndrome, causado por mutaciones en FOXF1, asocia con frecuencia otras malformaciones, incluida atresia de esófago y fistula T-E (Stankiewicz et al., 2009).
Entre las enfermedades monogénicas que presentan con frecuencia AE, cabe destacar el síndrome AEG (Anoftalmia-Esófago-Genital), el síndrome de Feingold y síndrome CHARGE (Tabla 1). Entre un 15 y 20% de AE, aparecerán asociadas a algún síndrome monogénico como los mencionados. También es esperable que la utilización de las técnicas de secuenciación de exoma resulten útiles, por una parte para identificar genes candidatos, y por otra, para aumentar la tasa de diagnóstico de síndromes monogénicos.
La asociación VACTERL, que no constituye por sí misma un síndrome como tal, sino un patrón definido de malformaciones que se repiten en diferentes pacientes sin que pueda atribuirse al mero azar, es una de la entidades que con más frecuencia asocia AE y siempre debe ser tenida en cuenta. Se han descrito diferentes microdeleciones en pacientes con asociación VACTERL, aunque no se ha podido establecer una relación efectiva con ninguna región concreta (Brosens et al., 2016). Un diagnóstico importante a tener en cuenta en casos de fenotipo VACTERL con defectos en eje radial, será la anemia de Fanconi (Tabla 1), un síndrome bien conocido, de herencia autosómico recesiva, y elevado riesgo de desarrollar importantes alteraciones hemato-oncológicas (Faivre et al., 2005).
Atresia de esófago compleja o sindrómica, relacionada con alteraciones ambientales
La AE también puede aparecer asociada a factores teratógenos, como ocurre en la embriopatía diabética y la ingesta de alcohol durante el embarazo (Martinez-Frias et al., 1991; Martinez-Frias et al., 1994), así como la exposición intraútero a ciertos fármacos, como el agente antitiroideo Metimazol (Gripp KW et al., 2011), y el Micofenolato de mofetilo (MMF), un inmunodepresor ampliamente utilizado en receptoras de trasplante de órganos y en enfermedades autoinmunes (Perez-Aytes et al., 2017). Estos factores siempre deberán ser tenidos en cuenta en la anamnesis de cualquier paciente con AE.
CONCLUSIÓN
La AE es una malformacion heterogénea, que puede aparecer asociada a múltiples entidades, tanto de origen genético como ambiental, lo que hace que la aproximación diagnóstica a un RN con AE suponga en muchas ocasiones un difícil reto, para lo cual proponemos un algoritmo diagnostico (Figura 2).
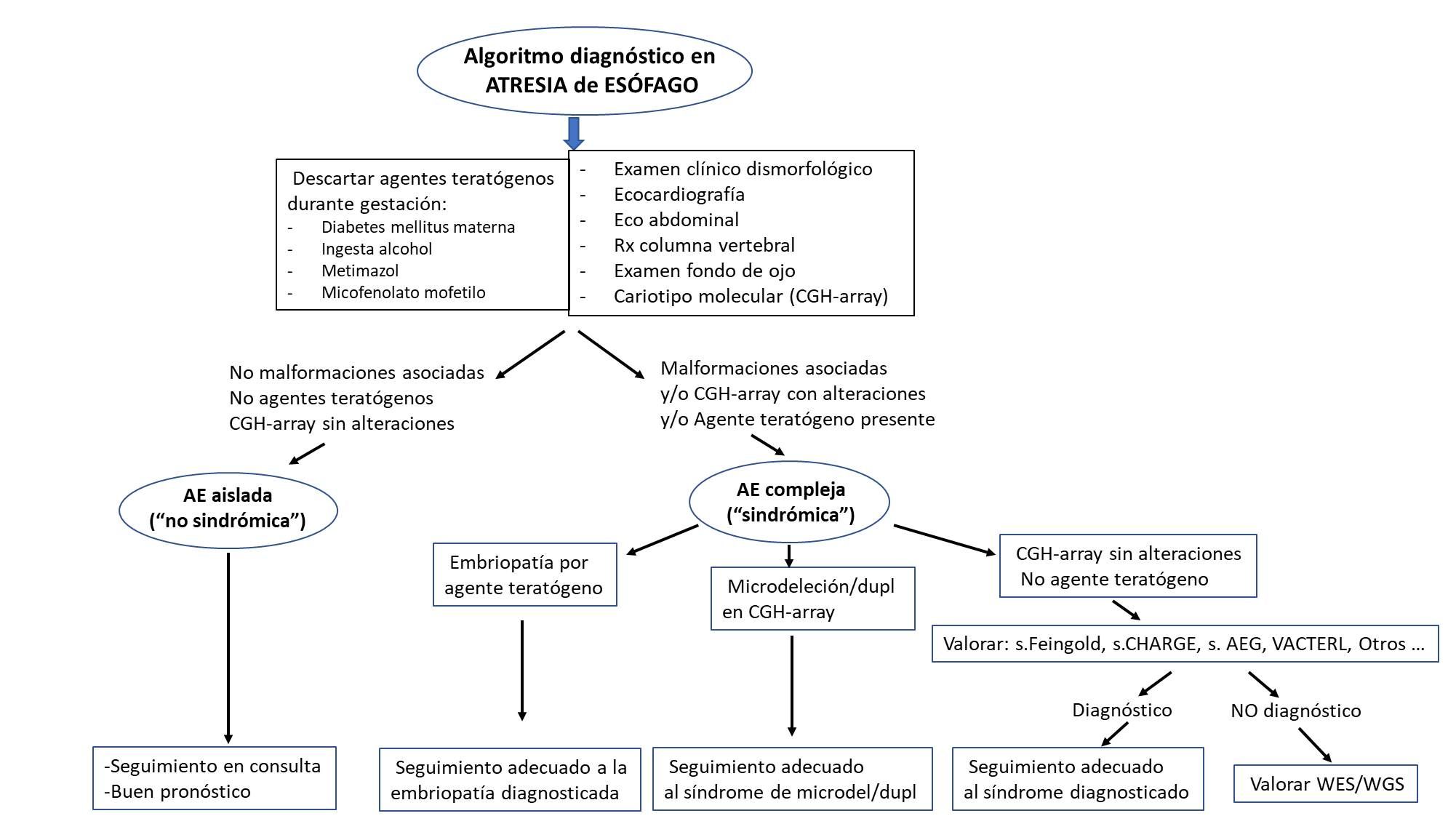
Siguiendo nuestro algoritmo, ante un RN con AE, deberíamos en primer lugar realizar una cuidadosa valoración clínica dismorfológica, así como tener en cuenta los antecedentes prenatales, considerando posibles agentes teratógenos, fundamentalmente diabetes mellitus materna, y exposición prenatal a alcohol, metimazol y MMF. Se realizará un despistaje de otras anomalías congénitas con ecocardiografía, ecografía cerebral y abdominal, radiografía vertebral y de extremidades y estudio de fondo de ojo. En todos los casos se recomienda la realización de un estudio de array-CGH, dado que algunas microdeleciones relacionadas con AE pueden presentar rasgos dismórficos muy inespecíficos en el período neonatal, y podrían identificarse inicialmente como AE aislada, aunque en casos en que la exploracion clínica impresione claramente de AE aislada, podría considerarse la opción de realizar un seguimiento clínico del paciente en lugar de solicitar estudios genéticos desde una primera instancia. Si no hay historia de exposición prenatal a potenciales teratógenos, las exploraciones complementarias no han detectado otras malformaciones y el array-CGH no muestra anomalías, la AE puede ser catalogada provisionalmente como no-sindrómica, siendo recomendable un seguimiento, como mínimo hasta el año de vida, para confirmar definitivamente que estamos ante una AE aislada.
En los casos de AE sindrómica, con malformaciones asociadas y/o rasgos dismórficos llamativos, si el array-CGH no detecta anomalías, los síndromes AEG, Feingold y CHARGE, y la asociacion VACTERL, son las principales entidades a tener en cuenta a la hora de indicar estudios de genética molecular. Tras completar el estudio, aquellos RN con AE sindrómica que queden sin un diagnóstico concreto, deberán llevar seguimiento en consulta para valorar la evolución del fenotipo y planificar eventuales estudios de genética molecular dirigidos a genes concretos, o plantearse estudios de exoma completo (WES) o incluso genoma (WGS) (Figura 2) .


