INTRODUCCIÓN
La muerte súbita cardíaca (MSC) se define como una muerte natural de causa cardíaca que ocurre en la primera hora des del inicio de los síntomas, en un paciente con o sin historia previa de enfermedad cardiovascular (Zipes et al., 1998). La MSC tiene una incidencia variable pero en general se encuentra en el rango de 50-100 casos por cada 100.000 personas en los países industrializados y es responsable del 80-90% de todas las muertes súbitas (MS) en adultos (Fishman et al., 2010). Al menos el 85% de las MSC son causadas por cardiopatías coronarias y afectan mayoritariamente a población adulta (mayor de 45 años) (Oliva 2011, Myerburg and Junttila 2012). Esta patología coronaria tiene una base genética mayoritariamente debida a la acumulación de variantes comunes, es decir, alteraciones genéticas presentes en más de un 1% de la población general.
El 20% de MSC restantes se deben a enfermedades cardíacas hereditarias debidas a variantes genéticas raras en la población (frecuencia menor del 1%) que suelen seguir un patrón de herencia mendeliano (mayoritariamente autosómico dominante). Aunque en la mayoría de casos las variantes genéticas son heredadas de sus progenitores existe la posibilidad de encontrar variantes de novo en el paciente como resultado de una mutación en una de las células germinales de los padres y por lo tanto, ningún antecedente la familia sería portador de ésta. También se han descrito casos de mosaicismo, dónde un individuo tiene dos o más poblaciones de células que difieren de su composición genética. Un ejemplo lo encontramos en dos gemelos afectados por el síndrome de Timothy y portadores de una mutación en el gen CACNA1C cuyos padres no presentan la mutación. La hipótesis del mosaicismo se confirmó tras la identificación de un pequeño pico de la mutación missense en DNA de la mucosa oral de la madre (Splawski et al., 2004).
Estas patologías suelen afectar a menores de 35 años y las podemos clasificar en 2 grandes grupos: las cardiomiopatías y las canalopatías.
Las cardiomiopatías se caracterizan por la presencia de alteraciones genéticas en genes que codifican para proteínas que forman parte del citoesqueleto del miocardio, así como del sarcómero, los desmosomas e incluso la membrana nuclear. Las principales son: cardiomiopatía hipertrófica, cardiomiopatía dilatada y la cardiomiopatía arritmogénica. Las canalopatías son un grupo heterogéneo de enfermedades genéticas causadas por la disfunción de los canales iónicos cardíacos y/o proteínas asociadas a estos. Las principales son el síndrome de QT largo (SQTL), el síndrome de Brugada (SBr), el síndrome de QT corto (SQTC) y la taquicardia ventricular polimórfica catecolaminérgica (TVPC). Todas las enfermedades cardíacas hereditarias (excepto la cardiomiopatía hipertrófica y recientemente también sugerida la dilatada son enfermedades raras, ya que afectan un porcentaje bajo de la población (menos de 1 en 2000) y se deben generalmente a variantes genéticas raras (McKenna et al., 2017).
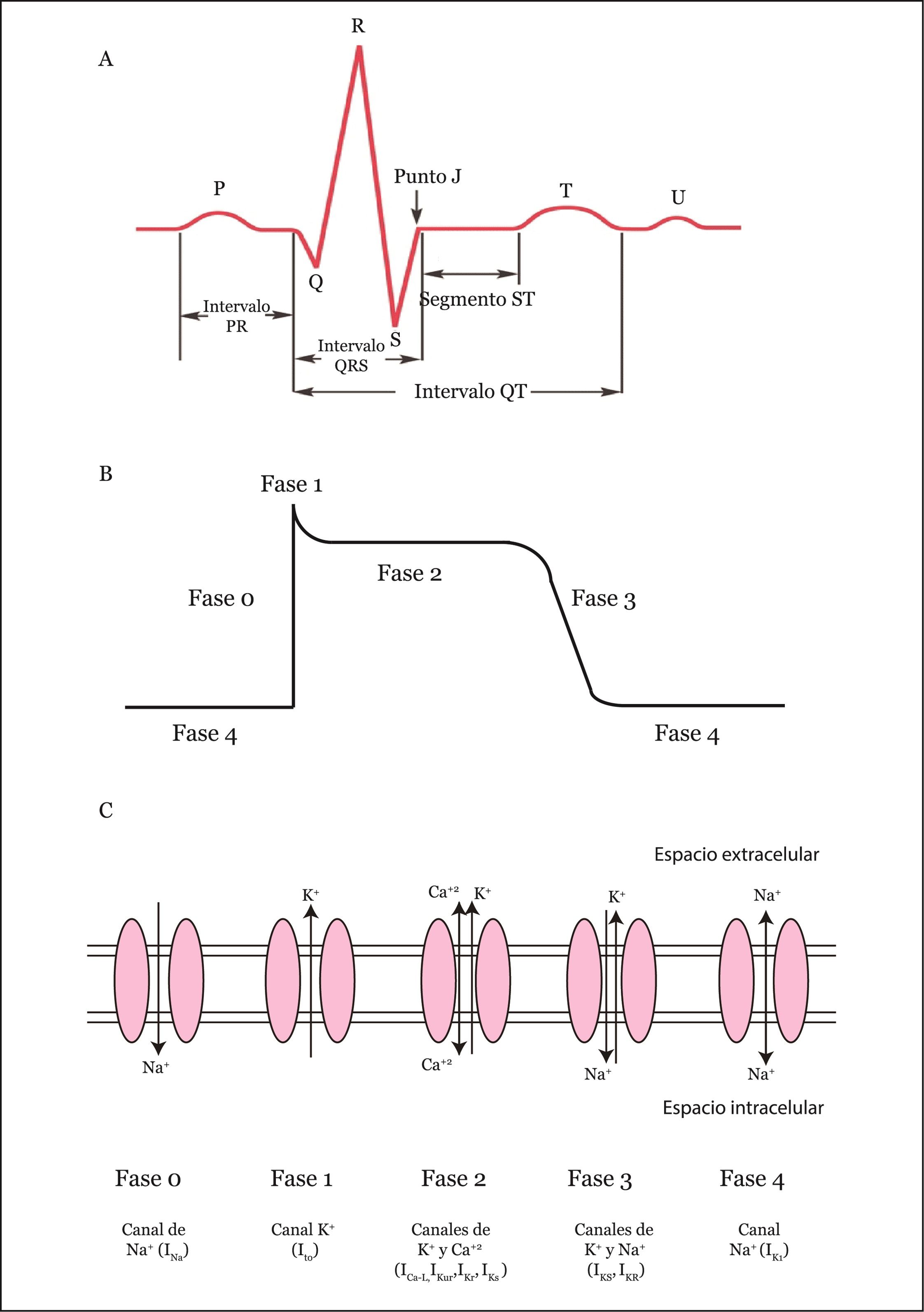
El electrocardiograma (ECG) es una representación en papel de los cambios eléctricos que se producen durante la transmisión de la señal eléctrica a lo largo del miocardio. Esta transmisión eléctrica es la responsable de la contracción del miocardio y consecuente bombeo de la sangre desde su interior hacia los vasos sanguíneos (Figura 1A). La actividad eléctrica del corazón es el resultado de un balance de corrientes de despolarización y repolarización y la dirección de las corrientes viene determinada por el gradiente electroquímico de cada ion (principalmente sodio, calcio y potasio). El balance del impulso eléctrico generado por este intercambio de iones en las células excitables se llama potencial de acción cardíaco (PAC). La forma típica de PAC ventricular consta de 5 fases (Figura 1B-C):
- Fase de despolarización rápida (o fase 0): en esta fase se produce una entrada masiva de iones de sodio a través de los canales de sodio dependientes de voltaje (INa). La diferencia de potencial de la membrana pasa de -90mV a +40mV.
- Repolarización parcial de la membrana (o fase 1): esta repolarización sutil se da por la inactivación de los canales de sodio y la apertura de los canales de potasio responsables del corriente transitorio de salida de potasio (Ito).
- Fase plateau (o fase 2): esta fase corresponde a la entrada del corriente de ICa por los canales de calcio tipo L (ICaL). Esta entrada de calcio activa los receptores de rianodina presentes en el retículo sarcoplasmático (RS) al citoplasma y permite la contracción de los cardiomiocitos.
- Fase de repolarización celular (o fase 3): el final de la fase 2 y la fase 3 corresponden al efecto de diferentes corrientes salientes de potasio.
- Fase de reposo (o fase 4): en esta fase los cardiomiocitos se mantienen estables en un potencial negativo de -90mV.
Enfermedades arritmogénicas hereditarias
El correcto funcionamiento del corazón se basa en la sincronización de las corrientes de entrada y salida de iones a través de los canales iónicos dependientes de voltaje que se encuentran en la membrana plasmática de los cardiomiocitos. Las variantes patogénicas en los genes que codifican para estos canales, o en sus proteínas asociadas, pueden resultar en malformaciones del canal que induzcan una alteración de sus propiedades biofísicas. Estos cambios pueden generar cambios en las corrientes y dar lugar a una fibrilación ventricular (FV) que lleve a una parada del bombeo cardiaco, a la pérdida de consciencia y a una posible MSC si no se recupera la correcta actividad eléctrica y el consecuente bombeo sanguíneo. Estas patologías arritmogénicas inducen cambios eléctricos en la transmisión de la señal eléctrica a través del corazón, pero sin inducir ningún tipo de malformación estructural. Esto puede provocar que la MSC sea la primera manifestación de la patología, sin síntoma previo. Por esta razón, la identificación precoz es crucial para poder prevenir episodios letales mediante tratamiento personalizado.
Penetrancia incompleta, expresividad variable y pleiotropía
Generalmente, las enfermedades cardíacas hereditarias siguen un patrón de herencia autosómico dominante pero como ocurre en otras enfermedades existe también el riesgo de encontrar penetrancia incompleta, expresividad variable y solapamiento genético entre enfermedades (Coll et al., 2017).
La penetrancia incompleta se define como la proporción de portadores de la alteración genética que no expresan el fenotipo. La expresividad variable se refiere al diferente grado de gravedad de la patología, así como la diferente edad de manifestación y evolución que pueden aparecer en diferentes pacientes con una misma variante genética. Tanto la penetrancia incompleta como la expresividad variable se dan probablemente por la combinación de factores genéticos, ambientales y del estilo de vida. En este tipo de enfermedades también encontramos solapamiento entre ellas, conocido con el término pleiotropía. Este fenómeno ocurre cuando variantes patogénicas en un único gen pueden dar como resultado diferentes enfermedades (Figura 2).
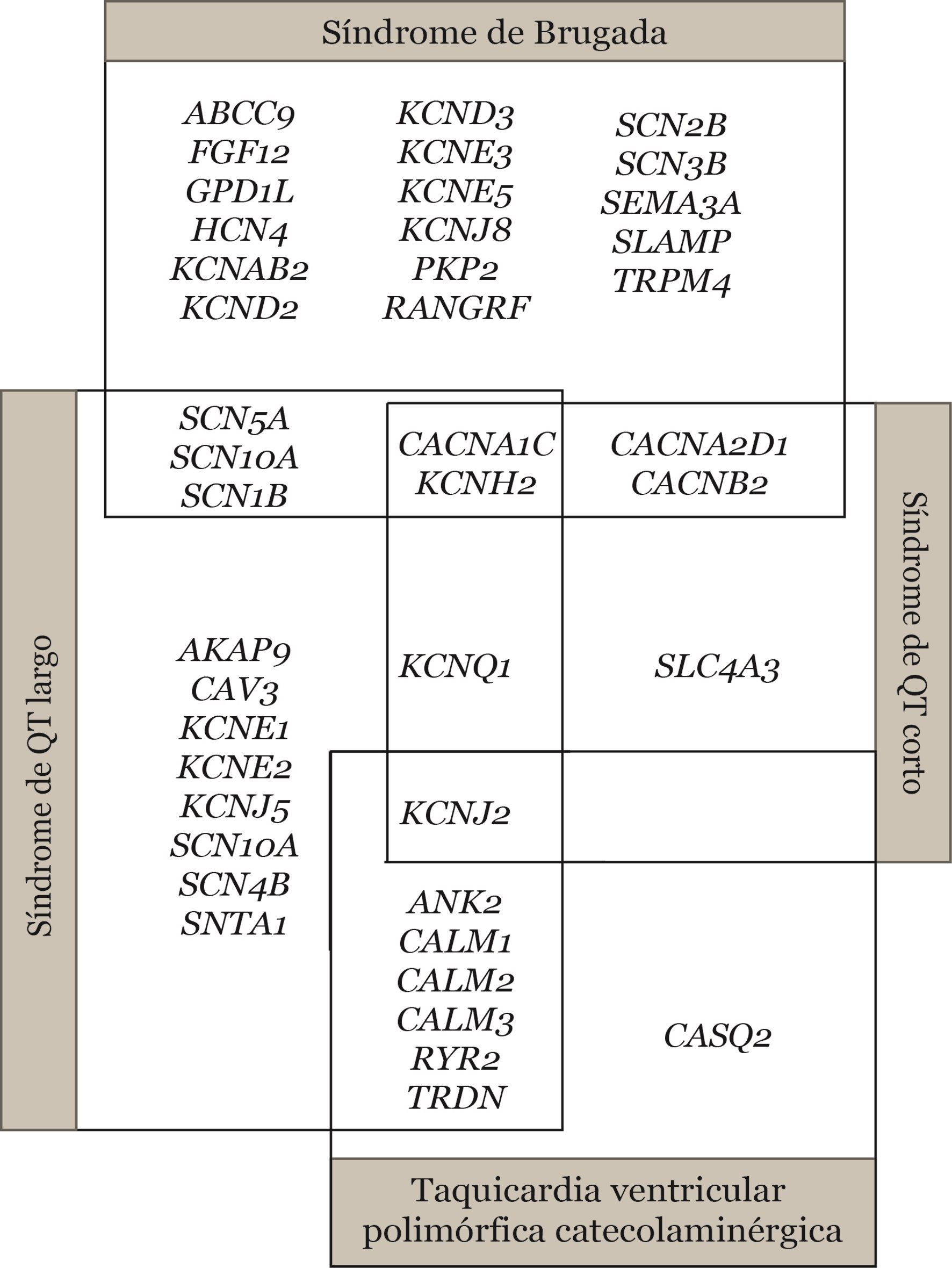
Dentro de los factores genéticos, encontramos la posibilidad de tener múltiples variantes raras (en un mismo gen o en genes distintos) con efecto aditivo, CNVs o nuevos genes asociados a la enfermedad aún por identificar. También existen estudios que asocian variantes comunes, llamadas “second hits”, que podrían estar modulando (positiva o negativamente) el efecto de otra variante rara. También existe una posibilidad poco estudiada como sería la eventualidad de mutaciones somáticas que afectaran de forma local, conocido también como “neural crest theory” (Brugada 2016).
Finalmente, podemos encontrar variantes en regiones no codificantes como las regiones intrónicas, UTR o en regiones que codifican para microRNAs. En los factores no genéticos encontramos la edad del paciente, el sexo y factores exógenos como la fiebre o la toma de determinados fármacos (Coll et al., 2017).
Estudio genético
La secuenciación automática Sanger ha sido y sigue siendo el método gold standard para la identificación de variantes genéticas. Sin embargo, el descubrimiento de una gran cantidad de nuevos genes asociados a las patologías puso en evidencia la necesidad de una secuenciación más rápida y económica. Esto ha llevado a las grandes compañías a desarrollar nuevas tecnologías para el análisis del genoma humano llamadas Next Generation Sequencing (NGS) o High Throughput Sequencing. Existen diferentes aproximaciones de secuenciación, como el diseño de paneles mediante sondas, en la que se analizan sólo determinados genes o regiones de interés; pero también existen aproximaciones más complejas como la secuenciación de exoma o incluso de genoma. En el campo de la cardiología y concretamente en las enfermedades cardíacas hereditarias el uso de paneles
es la primera de las opciones a tener en cuenta para de estudiar las causas genéticas de la enfermedad. Esta aproximación nos permite identificar variantes en un único nucleótido (SNPs), pequeñas inserciones-deleciones (indels) y variantes en el número de copias (CNVs) ya sean todas ellas variantes comunes (frecuencia del alelo menor >1%), raras (frecuencia del alelo menor <1%) o privadas. Sin embargo, el diagnóstico genético positivo oscila del 30% en pacientes con SBr al 85% en pacientes con SQTL, quedando muchos casos sin resolver genéticamente. La secuenciación de exoma o genoma sería una opción a tener en cuenta en aquellos casos sin resolver ya que con ella se pueden identificar nuevos genes y/o regiones asociadas a la patología.
Síndrome de Brugada
El SBr fue descrito por primera vez en 1992 por los hermanos Brugada (Brugada y Brugada 1992). En su representación en el ECG se caracteriza por la elevación del segmento ST en las derivaciones V1-V3 que induce anomalías en la conducción eléctrica cardíaca en pacientes sin alteraciones estructurales llevando a la predisposición para presentar arritmias ventriculares y MSC (Benito 2009). Existen 3 patrones electrocardiográficos diferentes de SBr pero sólo el tipo I es considerado diagnóstico de la enfermedad (Wilde et al., 2002, Antzelevitch et al., 2005).
Tiene una prevalencia estimada de 1-5/10.000 habitantes en Europa aunque ésta puede estarse subestimando debido a las formas silenciosas de la enfermedad (Berne y Brugada 2012). Sin embargo, el SBr parece tener una prevalencia menor en el norte de Europa (1.1/100.000) y superior en el Sud-Este de Asia (12/10.000) (Holst et al., 2012, Nademanee et al., 1997). El SBr está clasificado como una enfermedad rara con un tipo de herencia autosómica dominante, con penetrancia incompleta y expresividad variable. La penetrancia del SBr depende de la edad y del sexo del paciente, siendo más letal en varones a partir de los 40 años de edad (3.34 veces mayor). Estas diferencias pueden estar relacionadas con el efecto diferencial de las hormonas sexuales en las densidades de corrientes en los canales iónicos (Juang and Horie et al., 2016).
Bases genéticas
En 1998, Chen et al describieron la primera alteración patogénica en el gen SCN5A (Chen et al., 1998). El gen SCN5A (localizado en 3p22.2) codifica para la subunidad alfa del canal de sodio cardíaco dependiente de voltaje (Nav1.5) y variantes patogénicas en este gen explican actualmente alrededor del 25% de los casos de SBr (Priori et al., 2015). Se han descrito más de 500 mutaciones en más de 20 genes diferentes que codifican para canales de sodio, calcio y potasio, así como proteínas que afectan y/o regulan estos canales. Las variantes patogénicas en todos estos genes adicionales pueden explicar hasta un 10% más de los casos, con lo que el 65% de los casos de SBr quedan genéticamente sin resolver tras un estudio genético exhaustivo de todos los genes conocidos hasta la fecha (Tabla 1) (Fernandez-Falgueras et al., 2017).
Tabla 1: Genes asociados a los diferentes tipos de Síndrome de Brugada.
| Gen | Corriente | Efecto funcional | Incidencia | Referencia |
| ABCC9 | IK-ATP | Ganancia de función | Rara | (Hu et al., 2014) |
| CACNA1C | ICaL | Perdida de función | 6-7% | (Antzelevitch et al., 2007) |
| CACNA2D1 | ICaL | Perdida de función | Rara | (Burashnikov et al., 2010) |
| CACNB2 | ICaL | Perdida de función | 4-5% | (Antzelevitch et al., 2007) |
| FGF12 | INa | Perdida de función | Rara | (Hennessey et al., 2013) |
| GPD1L | INa | Perdida de función | Rara | (Makiyama et al., 2008) |
| HCN4 | If | Ganancia de función | Rara | (Crotti et al., 2012) |
| KCNAB2 | Ito | Ganancia de función | Rara | (Portero et al., 2016) |
| KCND2 | Ito | Ganancia de función | Rara | (Perrin et al., 2014) |
| KCND3 | Ito | Ganancia de función | Rara | (Giudicessi et al., 2011) |
| KCNE3 | Ito / IKs | Perdida de función | Rara | (Delpon et al., 2008) |
| KCNE5 | Ito / IKs | Ganancia de función | Rara | (Ohno et al., 2011) |
| KCNH2 | IKr | Ganancia de función | Rara | (Verkerk et al., 2005) |
| KCNJ8 | IK-ATP | Ganancia de función | Rara | (Medeiros-Domingo et al., 2010) |
| LRRC10 | – | – | Rara | (Huang et al., 2017) |
| PKP2 | – | Perdida de función | Rara | (Cerrone et al., 2014) |
| RANGRF | INa | Perdida de función | Rara | (Kattygnarath et al., 2011) |
| SCN1B | INa | Perdida de función | 1-2% | (Watanabe et al., 2008) |
| SCN2B | INa | Perdida de función | Rara | (Riuro et al., 2013) |
| SCN3B | INa | Perdida de función | Rara | (Hu et al., 2009) |
| SCN5A | INa | Perdida de función | 11-24% | (Chen et al., 1998) |
| SCN10A | INa | Perdida de función | 2-5% | (Hu et al., 2014) |
| SEMA3A | Ito | Ganancia de función | Rara | (Boczek et al., 2014) |
| SLMAP | INa | Perdida de función | Rara | (Ishikawa et al., 2012) |
| TRMP4 | NSCCa | Perdida/ganancia de función | Rara | (Liu et al., 2013) |
Variantes patogénicas en genes que codifican para las subunidades β del canal de sodio cardiaco como SCN1B, SCN2B y SCN3B pueden modificar también la función del canal. El gen SCN1B codifica para las subunidades β1 y β1b las cuales son proteínas auxiliares modificadoras de la función del canal. Mutaciones en este gen provocan una disminución de la corriente de sodio ya que tiene efecto en el transporte del canal de sodio cardíaco a membrana (Watanabe et al., 2008). Los genes SCN2B y SCN3B codifican para las subunidades β2 y β3, respectivamente, del canal de sodio cardíaco y mutaciones en ambos llevan a una pérdida de función del canal de sodio cardíaco (Riuro et al., 2013, Hu et al., 2009). También se han descrito mutaciones asociadas al SBr en el canal de sodio neuronal SCN10A, que modula la expresión de SCN5A y la función eléctrica del corazón (Hu et al., 2014). Se han identificado mutaciones en genes que pueden regular la expresión y/o la localización de los canales de sodio,por ejemplo, en los genes RANGRF, GPD1L, SLAMP, PKP2 y TRPM4. A parte de genes asociados a los canales de sodio, también se han descrito mutaciones asociadas al SBr en genes que codifican para canales de calcio y potasio. En el caso de los canales de calcio encontramos los genes CACNA1C, CACN1B y CACNA2D1. Se detectan mutaciones en CACNA1C y CACN1B en un 11.5% de los pacientes con SBr (Kapplinger et al., 2010). Además, se encuentran también mutaciones que producen ganancia de función en genes que codifican para canales de potasio como KCNAB2, KCND3, KCNE3, KCNE5 y KCNJ8.
Hasta el momento, en la genética del SBr tan solo se habían considerado las variantes puntuales o pequeñas inserciones o deleciones. Sin embargo, en 2011 Eastaugh et al identificó la primera variante en número de copia que implicaba la deleción de los exones 9 y 10 del gen SCN5A (Eastaugh et al., 2011). Aunque la llegada de las nuevas tecnologías facilita la identificación de este tipo de reordenamientos el porcentaje de variantes de este tipo en el gen SCN5A y en los genes minoritarios sigue siendo bajo. (Mademont-Soler et al., 2016)
Síndrome de QT Largo
El SQTL es un desorden eléctrico cardíaco caracterizado por una prolongación anormal del intervalo QT en el ECG. Actualmente se considera un valor de QTc (QT corregido respecto a la frecuencia cardiaca) mayor de 480 ms como diagnóstico en ambos sexos (o bien un score Schwartz ≥3) (Goldenberg and Moss 2008). Los pacientes con SQTL pueden presentar arritmias ventriculares como torsade de pointes y MSC. La prevalencia estimada del SQTL es de 1 en 2000 individuos (Schwartz et al., 2009). Aunque la mayoría de los pacientes pueden estar sin diagnosticar durante su vida, hasta un 13% puede sufrir una MSC y un 36% sufre algún sincope antes de los 40 años de edad (Priori et al., 2003). Evitar los medicamentos que potencialmente pueden prolongar el QT es una recomendación universal para todos los pacientes con SQTL.
Bases genéticas
El SQTL se caracteriza por una prolongación del PAC debido al incremento de corrientes entrantes (principalmente de sodio y calcio) o la disminución de corrientes salientes de potasio. La mayoría de mutaciones descritas siguen un patrón de herencia autosómica dominante excepto el síndrome de Jervell y Lange-Nielsen que sigue un patrón de herencia autosómica recesiva (Tabla 2). Aproximadamente el 75% de los pacientes con SQTL presentan alteraciones patogénicas en alguno de los 3 genes principales: KCNQ1 (SQTL1), KCNH2 (SQTL2) o SCN5A (SQTL3). El SQTL1 causado por variantes en el gen KCNQ1 recoge aproximadamente un 35% de los casos de SQTL. Este gen codifica para la proteína Kv7.1 del canal de K+ dependiente de voltaje, proteína fundamental en la fase de repolarización del potencial de acción. Las variantes patogénicas identificadas en este gen producen una pérdida de función del canal y una disminución de la corriente IKs, que explica la prolongación del potencial de acción y el intervalo QT en el ECG. El STQL2 se da en un 25% de los casos y está causado por variantes patogénicas en el gen KCNH2, también denominado HERG. La proteína que codifica, Kv11.1, es responsable de la activación rápida IKr. Finalmente, el STQL3 es causado por variantes patogénicas en el gen SCN5A y puede llegar a explicar el 15% de los casos de SQTL. Alteraciones en otros genes minoritarios que codifican para otros canales iónicos cardíacos o proteínas que interaccionan con estos canales y que regulan la función del canal pueden llegar a explicar hasta un 10% adicional de casos, quedando un 15% de casos sin causa genética tras un estudio genético exhaustivo (Ackerman et al., 2013). La mayoría de variantes patogénicas asociadas al SQTL son substituciones de un único nucleótido y pequeñas inserciones/deleciones dentro de las regiones codificantes. Sin embargo, también se han descrito reordenamientos genéticos que resultan en grandes deleciones o duplicaciones del gen (Campuzano et al., 2014).
Tabla 2: Genes asociados al Síndrome de QT largo.
| Gen | Corriente | Efecto funcional | Incidencia | Referencia |
| AKAP9 | IKs | Perdida de función | Rara (SQTL11) | (Chen et al., 2007) |
| ANK2 | ICaL | Alteración localización canales | Rara (SQTL4) | (Mohler et al., 2003) |
| CACNA1C | ICaL | Ganancia de función | Rara (Síndrome de Timothy, SQTL8) | (Boczek et al., 2013) |
| CALM1 | ICaL | Múltiples | Rara (SQTL14) | (Crotti et al., 2013) |
| CALM2 | ICaL | Rara (SQTL15) | (Crotti et al., 2013) | |
| CALM3 | ICaL | Rara (SQTL16) | (Reed et al., 2015) | |
| CAV3 | ICa-T | Ganancia de función | Rara (SQTL9) | (Vatta et al., 2006) |
| KCNE1 | IKs | Perdida de función | Rara (SQTL5) | (Splawski et al., 1997) |
| KCNE2 | IKr | Perdida de función | Rara (SQTL6) | (Splawski et al., 2000) |
| KCNH2 | IKr | Perdida de función | 25% (SQTL2) | (Curran et al., 1995) |
| KCNJ2 | IK1 | Perdida de función | Rara (Síndrome de Andersen-Tawil, SQTL7) | (Tristani-Firouzi et al., 2002) |
| KCNJ5 | IK,Ach | Perdida de función | Rara (SQTL13) | (Yang et al., 2010) |
| KCNQ1 | IKs | Perdida de función | 35% (SQTL1) | (Mannens y Wilde 1997) |
| RYR2 | ICa | Ganancia de función | Rara | (Kauferstein et al., 2011) |
| SCN10A | INa | Perdida de función | Rara | (Abou Ziki et al., 2017) |
| SCN1B | INa | Ganancia de función | Rara | (Lopez-Santiago et al., 2007) |
| SCN4B | INa | Ganancia de función | Rara (SQTL10) | (Medeiros-Domingo et al., 2007) |
| SCN5A | INa | Ganancia de función | 15% (SQTL3) | (Wang et al., 1995) |
| SNTA1 | INa | Ganancia de función | Rara (SQTL12) | (Ueda et al., 2008) |
| TRDN | ICaL | Ganancia de función | Rara | (Altmann et al., 2015) |
Síndrome de QT Corto
El SQTC fue descrito por primera vez en el año 2000 (Gussak et al., 2000). Esta canalopatía se caracteriza por un acortamiento del intervalo QT en el ECG y una predisposición a desarrollar arritmias ventriculares. Actualmente se considera un valor de QTc (QT corregido) menor de <350 ms como diagnóstico (Monteforte 2012). Es una enfermedad muy rara y, por lo tanto, hay muy pocos datos al respeto. La parada cardíaca es el síntoma más común (40% de los pacientes) y es más predominante en varones con una media de edad de 26±15 años (Giudicessi y Ackerman 2013).
Bases genéticas
El SQTC es una enfermedad genética hereditaria pero en sólo el 15% de los casos se identifica una variante posiblemente causal (Garcia-Elias y Benito 2018). Hasta el momento se han descrito alteraciones patogénicas en 7 genes diferentes que siguen un patrón de herencia autosómica dominante. Se demuestra así la elevada penetrancia y siendo responsable del 60% de los casos (Tabla 3). Todas ellas afectan a genes que codifican para canales de calcio y potasio. El subtipo más prevalente está asociado a variantes patogénicas con ganancia de función en el gen KCNH2 (SQTC1) que aumentan la corriente a través del canal y acorta la duración del PAC y el intervalo QT (Brugada et al., 2004). El SQTC tipo 2 está asociado a variantes patogénicas en el gen KCNQ1 el cual produce un aumento de la función de los canales de potasio produciendo un acortamiento del PAC. El síndrome de QT corto tipo 3 es causado por variantes patogénicas en el gen KCNJ2, produciendo una aceleración de la fase 3 del PAC (Campuzano et al., 2015). Se han descrito también muchas variantes patogénicas que producen pérdida de función en genes que codifican para diferentes subunidades. Entre ellos encontramos los canales de calcio como CACNA1C, CACNB2 y CACNA2D1. El gen CACNA1C codifica para la proteína CaV1.2 y se ha visto que las variantes patogénicas identificadas en este gen producen un acortamiento del PAC reduciendo el tráfico de la subunidad α1C a membrana (Antzelevitch et al., 2007). El gen CACNB2 codifica para la subunidad β de los canales de calcio dependientes de voltaje tipo L y las variantes patogénicas en él producen una disminución del corriente ICaL pero no afectan al tráfico del canal (Garcia-Elias y Benito 2018). Finalmente, en 2011 Templin C. et al reportaron por primera vez una alteración en el gen CACNA2D1 que causaba SQTC (Templin et al., 2011). Este gen codifica para la subunidad α2δ1 de los canales de calcio dependientes de voltaje tipo L. Recientemente se ha identificado una variante patogénica missense en dos familias afectadas por el SQTC en el gen SLC4A3 (Thorsen et al., 2017). Esta alteración reduce la expresión de AE3 y disminuye el transporte de bicarbonato a la membrana. El análisis mecanístico sugiere que un incremento en el pH y una disminución de Cl- acortarían la duración del potencial de acción cardíaco.
Tabla 3: Genes asociados al Síndrome de QT corto
| Gen | Corriente | Efecto funcional | Incidencia | Referencia |
| CACNA1C | ICaL | Perdida de función | Rara | (Antzelevitch et al., 2007) |
| CACNA2D1 | ICaL | Perdida de función | Rara | (Templin et al., 2011) |
| CACNB2 | ICaL | Perdida de función | Rara | (Antzelevitch et al., 2007) |
| KCNH2 | IKr | Ganancia de función | 15 % (SQTC1) | (Brugada et al., 2004) |
| KCNJ2 | IK1 | Ganancia de función | Rara (SQTC3) | (Priori et al., 2005) |
| KCNQ1 | IKs | Ganancia de función | Rara (SQTC2) | (Bellocq et al., 2004) |
| SLC4A3 | – | Cambios fisicoquímicos | Rara | (Thorsen et al., 2017) |
Taquicardia ventricular polimórfica catecolaminérgica
La TVPC se caracteriza por episodios de sincopes durante el ejercicio o ante fuertes emociones en individuos sin anomalías cardíacas estructurales (Napolitano et al., 1993). Aunque es una enfermedad rara, con una prevalencia estimada de 1 en 10.000, se trata de una enfermedad extremadamente severa (Napolitano et al., 2014). El ECG basal no suele mostrar alteraciones con lo que es necesario un ECG de 24 horas, una prueba de esfuerzo y el test Epinefrina o Isoproterenol para su diagnóstico (Lopez-Perez et al., 2014). La base molecular de la TVPC se encuentra
en una liberación anómala del calcio proveniente del RS en respuesta a la estimulación adrenérgica.
Bases genéticas
En 1999 Swan et al estudiaron 2 familias con TVPC y asignaron el locus de la enfermedad en el cromosoma 1q42-q43 (Swan et al., 1999). Posteriormente, Priori et al demostró que el gen asociado con la TVPC era el gen de la rianodina (RYR2) (Priori et al., 2001). Un 50-60% de los pacientes son portadores de una variante patogénica en este gen. El gen RYR2 codifica para un canal de calcio intracelular necesario para la excitación-contracción de los cardiomiocitos (Priori et al., 2001). Los receptores cardíacos de rianodina son canales de liberación de calcio presentes en el RS, un orgánulo intracelular que juega un papel clave en la regulación de la homeóstasis del Ca2+ en el corazón. El calcio se une al receptor RyR2 y desencadena la apertura de los canales permitiendo una salida rápida de Ca2+ del RS. Esta elevada concentración de Ca2+ citoplasmático induce una contracción del miocardio. Este ciclo está regulado muy finamente y una disfunción en él se asocia a enfermedades cardíacas como la TVPC (Leenhardt et al., 2012).
Poco después, Lahat et al identificaron un locus en el cromosoma 1p13-21 que se heredaba de forma recesiva y se detectó una variante patogénica missense en una región altamente conservada del gen CASQ2, donde se encuentra el mayor reservorio de calcio del RS de los cardiomiocitos (Lahat et al., 2001). Un 5% de los pacientes con TVPC presentan variantes patogénicas en este gen y en menor porcentaje en otros genes como las calmodulinas, ANK2 y KCNJ2 (Tabla 4).
Tabla 4: Genes asociados a la taquicardia ventricular polimórfica catecolaminérgica. RS: retículo sarcoplasmático.
| Gen | Efecto funcional | Incidencia | Referencia |
| ANK2 | Perdida de función | <1% | (Mohler et al., 2004) |
| CALM1 | Liberación Ca2+ del RS debido a pérdida de interacción CaM-RyR2 | <1% | (Nyegaard et al., 2012) |
| CALM2 | Reducción afinidad unión al Ca2+ en el dominio CaM | <1% | (Makita et al., 2014) |
| CALM3 | Reducción afinidad unión al Ca2+ en el dominio CaM | <1% | (Gomez-Hurtado et al., 2016) |
| CASQ2 | Disminución del contenido en Ca2+ en el RS | 2-5% | (Lahat et al., 2001) |
| KCNJ2 | Perdida de función | <1% | (Vega et al., 2009) |
| RYR2 | Saturación Ca2+ citoplasmático debido a liberación Ca2+ del RS | 50-60% | (Priori et al., 2001) |
| TRDN | Saturación de Ca2+ citoplasmático debido a liberación Ca2+ del RS | <1% | (Roux-Buisson et al., 2012) |
Conclusiones
Las enfermedades cardíacas hereditarias siguen mayoritariamente un patrón de herencia autosómico dominante. Sin embargo, como ocurre en otras enfermedades mendelianas, presentan penetrancia incompleta, expresividad variable y solapamiento fenotípico con otras enfermedades. La utilización de la secuenciación masiva en la práctica clínica se está convirtiendo por su coste-efectividad en una herramienta clave en el diagnóstico genético de las enfermedades cardíacas hereditarias. La identificación de una causa genética de la enfermedad puede ayudar especialmente a otros miembros de la familia que pueden estar en riesgo de sufrir una arritmia maligna con un posible desenlace letal.
CONFLICTO DE INTERESES
Ninguno.
AGRADECIMIENTOS
Obra Social ‘La Caixa’, Fondo de Investigación Sanitaria (FIS, PI14/01773) del Instituto de Salud Carlos III (ISCIII), y “Fundació Privada Daniel Bravo Andreu”. CIBERCV es una iniciativa del ISCIII, Ministerio de Economía y Competitividad de España.


