INTRODUCCIÓN
La leucemia mieloblástica aguda (LMA) es una neoplasia maligna que se caracteriza por la acumulación de blastos mieloides inmaduros (más del 20%) que reemplazan a la hematopoyesis normal en la médula ósea y sangre periférica de los pacientes afectados (Dohner et al., 2015; Sanz et al., 2016; Szer, 2012; Tallman et al., 2005). Los tratamientos actuales consiguen tasas de supervivencia a 5 años inferiores al 30% en pacientes mayores de 65 años y en torno al 40% en pacientes más jóvenes (Almeida y Ramos, 2016; Bryan y Jabbour, 2015; Sanford y Ravandi, 2015; Saraceni et al., 2016). Las repeticiones internas en tándem (internal tandem duplications, ITD) en el gen FLT3 (FMS-like tirosine kinase-3) constituyen la variante más frecuente en los pacientes con LMA, con una frecuencia entre el 20 y el 30% de los casos (Nakao et al., 1996).
Aunque la LMA se considera habitualmente una enfermedad agresiva, existe una considerable heterogeneidad en cuanto a la genética, la evolución clínica y el pronóstico de los pacientes con esta enfermedad (Arber et al., 2016; Dohner et al., 2017; Pulte et al., 2016). En las últimas décadas, se han identificado anomalías cromosómicas recurrentes y variantes genéticas, que constituyen la base para la clasificación y la estratificación de riesgo de la LMA (Arber et al., 2016; Vardiman et al., 2009). La última revisión (en 2017) de las recomendaciones del diagnóstico y manejo de la LMA de la ELN (European Leukemia Net) categoriza a los pacientes en tres grupos de riesgo basándose en las anomalías citogenéticas y moleculares (Dohner et al., 2017). Según esta revisión, el grupo de riesgo favorable incluye aquellas LMAs que presentan alguno de estos cuatro escenarios: translocación t(15;17)/PML‐RARα (Leucemia Promielocítica Aguda, LPA); reordenamientos que involucran a los factores de transcripción core binding factor(CBF), como la t(8;21)/RUNX1‐RUNX1T1 y la inv(16) o t(16;16)/CBFb‐MYH11; cariotipo normal con mutaciones bialélicas en el gen CEBPA; y cariotipo normal con mutaciones en el gen NPM1en ausencia de la duplicación interna en tándem de FLT3 (FLT3‐ITD). En la mencionada última versión de esta clasificación, se ha incluido además el valor de la ratio “alelo-ITD/alelo-WT” de FLT3, considerando que esta mutación únicamente tiene valor pronóstico adverso en caso de encontrarse en ratios iguales o superiores a 0,5. Sin embargo, este criterio ha sido puesto en duda por algunos autores, que defienden que es suficiente con la presencia de la mutación para excluir a un paciente con LMA del subgrupo de bajo riesgo, tal como se refleja en clasificaciones anteriores y en otros sistemas de estratificación similares, como el americano (National Comprehensive Cancer Network, NCCN) (Dohner et al., 2010; O’Donnell et al., 2017; Straube et al., 2018).
En la actualidad hay aprobados dos fármacos dirigidos para los pacientes positivos tanto para FLT3‐ITD como para FLT3-TKD (mutaciones en el dominio tirosin-quinasa): Rydapt (Midostaurina) para el momento de diagnóstico, y Xospata (Gilteritinib) para pacientes resistentes o en recaída, y se están desarrollando otros inhibidores de FLT3 que están en fases finales de ensayo clínico, pendientes de aprobación, como Crenolanib, Quizartinib y Sorafenib, aunque hay muchas otras moléculas en desarrollo (Tabla 1) (Patnaik, 2018; Rasko y Hughes, 2017; Thomas y Campbell, 2018; Wu et al., 2018). Los fármacos están aprobados para pacientes portadores de las mencionadas mutaciones, independientemente de la citogenética de la enfermedad, por lo que la determinación genética de FLT3 ahora no es dependiente de los resultados de cariotipo. Por tanto, la determinación de las variantes genéticas de FLT3 ya no conllevan únicamente un valor pronóstico para un determinado subgrupo de LMA, sino que la disponibilidad de medicamentos dirigidos les confieren un valor predictivo para todos los casos de LMA, lo que exige más que nunca un correcto diagnóstico genético de las alteraciones en este gen.
| Fármaco | IC50 FLT3 | Otras dianas | |||
| ITD | D835Y | ITD/D835Y | ITD/F691L | ||
| Fármacos aprobados | |||||
| Gilteritinib (ASP2215) | 2 | 2 | 2 | 22 | AXL, LTK |
| Midostaurina (PKC412, CGP41251) | 8 | <10 | 15 | 10 | KIT, PDGFR, PKC, VEGFR2 |
| Fármacos en últimas fases de ensayo clínico | |||||
| Crenolanib (CP-868-596) | 9 | 5 | 12 | 55 | KIT, PDGFR |
| Quizartinib (AC220) | <1 | 6 | 23-35 | 128 | KIT, PDGFR |
| Sorafenib | 1-2 | >1500 | >2000 | >2300 | KIT, PDGFR, RAF, VEGFR2/3, ERK |
Tabla 1. Fármacos con actividad contra FLT3. Los compuestos señalados en negrita están ya aprobados por la FDA. Otros fármacos que están en desarrollo en estos momentos son: AMG 925, Cabozantinib, FF-10101, G-749, Lestaurtinib (CEP-701), SEL24-B489, Sunitinib (SU11248), Tandutinib (MLN518, CT53518), y TTT-3002. Modificada de Patnaik (Patnaik, 2018) y Wu et al. (Wu et al., 2018).
IC50: concentración de inhibición al 50% de la proliferación de células Ba/F3 transformadas con plásmidos portadores de la/s variante/variantes de FLT3 indicadas.
Variantes genéticas del gen FLT3
El gen FLT3, también llamado quinasa hepática fetal 2 (FLK2), quinasa de células madre (STK-1) o clúster de diferenciación antigénica 135 (CD135), se encuentra en el brazo largo del cromosoma 13 (13q12.2), y tiene una longitud de 97,3 kb. La proteína codificada es un receptor de membrana que está formado por un dominio extracelular -compuesto por cinco dominios tipo inmunoglobulina-, una región transmembrana, y dos dominios tirosina-quinasa citoplasmáticos (Figura 1). En presencia de ligando, el receptor se activa y dimeriza, lo que conduce a su autofosforilación y fosforilación adicional de múltiples moléculas efectoras citoplasmáticas. Este gen participa en la hematopoyesis, principalmente en la diferenciación de las células precursoras, la proliferación de éstas y su supervivencia. En condiciones normales, FLT3 se expresa en los tejidos en los que se forman las células precursoras, como la médula ósea, los órganos linfoides, el hígado, el timo y la placenta (Meshinchi y Appelbaum, 2009); se expresa en todas las células hematopoyéticas CD34+, en los progenitores de linfocitos B, en los progenitores de la estirpe mieloide y en los monocitos. Aquellas células progenitoras que expresen en la membrana altos niveles del receptor se diferenciarán a macrófagos, mientras que las que expresen niveles más bajos serán eritrocitos (Grafone et al., 2012).
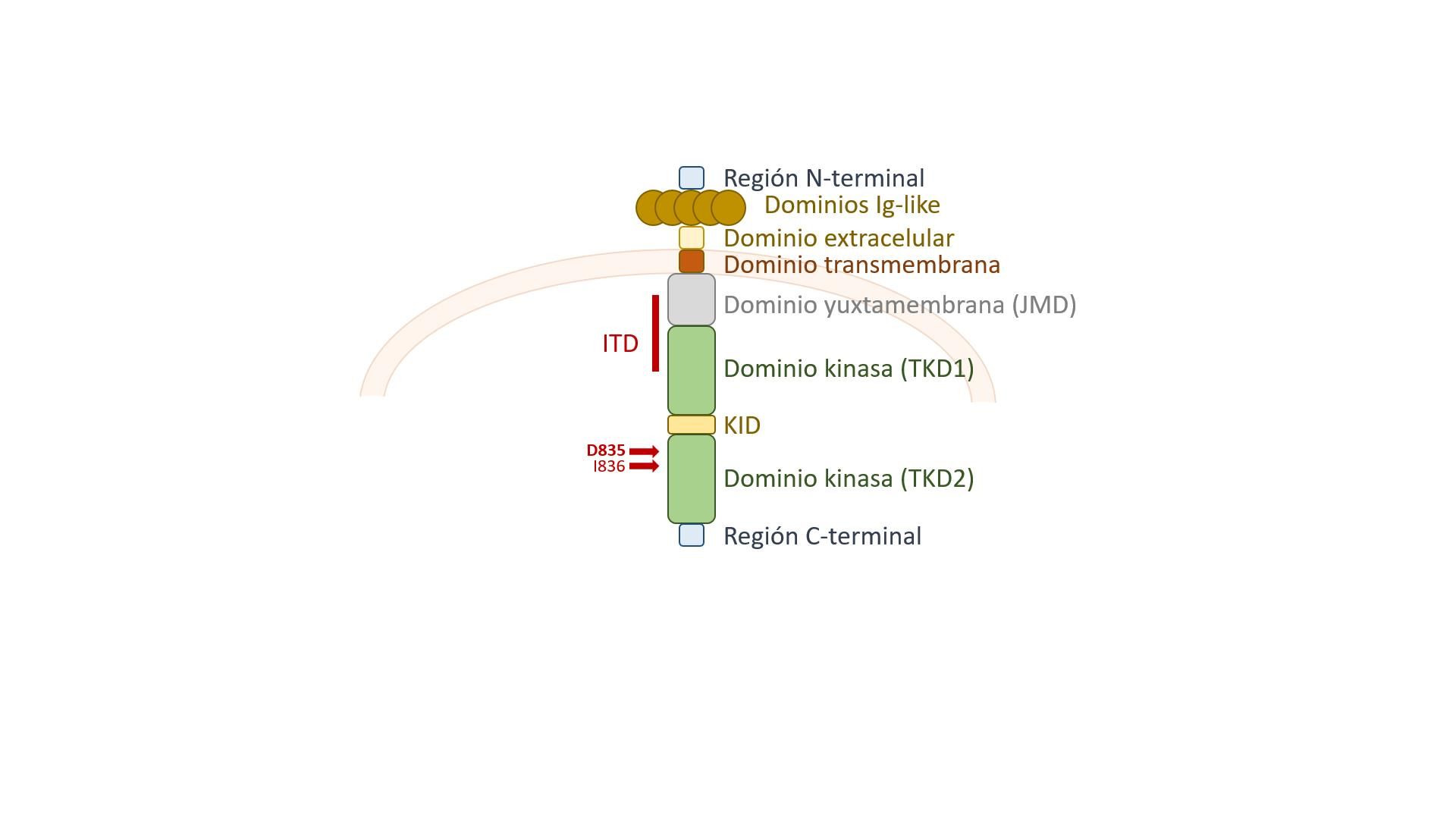
Figura 1. Estructura del receptor FLT3, y localización de las variantes más frecuentes. Las inserciones de duplicación en tándem (ITD) se localizan en los dominios yuxtamembrana (JMD) y quinasa 1 (TKD1), mientras que las variantes puntuales más frecuentes se localizan en el dominio quinasa 2 (TKD2). También se han publicado algunas variantes puntuales mucho menos frecuentes a lo largo de toda la proteína, desde el dominio extracelular (Patnaik, 2018).
Las mutaciones ITD en el gen FLT3 consisten en la inserción de secuencias repetidas: la mayoría (70%) se insertan en el dominio yuxtamembrana (JMD), mientras que el resto se localizan en el dominio tirosina-quinasa 1 (TKD1) (Figura 1) (Breitenbuecher et al., 2009; Kayser et al., 2009). Las variantes TKD1-ITDs son significativamente más largas que las JMD-ITD y se asocian a peor pronóstico (Arreba-Tutusaus et al., 2016; Kayser et al., 2009; Schlenk et al., 2014). Estudios estructurales han demostrado que tan sólo 15 pares de bases de inserción ITD en JMD pueden impedir la conformación auto-inhibitoria de la quinasa, permitiendo así que el receptor FLT3 se dimerice independientemente de la presencia de ligando, lo que conlleva auto-fosforilación constitutiva y activación de la señalización de vías de proliferación y supervivencia celular (Chan, 2011; Griffith et al., 2004). Se han reportado algunos casos (poco frecuentes) de deleciones, que también provocan activación constitutiva del receptor y, por tanto, tienen un efecto patogénico, al igual que las ITD (Chatain et al., 2015).
Otro tipo de alteraciones frecuentes encontradas en FLT3 son las mutaciones puntuales en el aminoácido Asp835 (D835), y las mutaciones puntuales e inserciones/deleciones en el aminoácido Ile836 (I836), situados en el dominio tirosina quinasa 2 (TKD2) (Figura 1). Estas mutaciones suelen denominarse genéricamente TKD, y suceden con una frecuencia en torno al 7% en los pacientes con LMA. La relación de las mutaciones TKD con el pronóstico está mucho más cuestionada (Pratz y Levis, 2017; Thiede et al., 2002), aunque ya se han desarrollado fármacos específicos para formas mutantes de FLT3 con estas aberraciones (Tabla 1).
La enorme relevancia clínica de la correcta caracterización molecular de las variantes del gen FLT3, en cuanto a la disponibilidad de tratamientos, y por la independencia de los resultados de cariotipo, ha conducido al desarrollo de distintas técnicas analíticas, que describimos a continuación.
MÉTODOS DE CARACTERIZACIÓN MOLECULAR DE FLT3
1. Detección de las variantes FLT3-ITD
Los métodos clásicos de detección de estas variantes se basan en la amplificación por PCR empleando cebadores específicos que flanquean la región donde se dan las variantes FLT3-ITD (exones 14 y 15), seguido del método elegido para visualizar la variante.
– Separación electroforética
Una vez amplificado el fragmento donde se encuentran las ITDs, el tamaño de los fragmentos obtenidos se puede visualizar en un gel de agarosa. Según el protocolo descrito por Nakao y colaboradores, el fragmento mutado puede presentar una longitud variable entre 134 y 366 pares de bases (Nakao et al., 1996).
Esta técnica presenta la ventaja de que es económica, fácil de interpretar, el resultado se obtiene con rapidez, es posible procesar un alto número de muestras simultáneamente, y el equipamiento que precisa está disponible en cualquier laboratorio de Biología Molecular. Sin embargo, esta técnica presenta el inconveniente de ser meramente cualitativa, esto es, no permite calcular la ratio “alelo-ITD/alelo-WT”. Además, la visualización del resultado precisa del uso de agentes intercalantes que presentan propiedades mutagénicas, lo que exige extremar las precauciones en su manipulación para minimizar riesgos, y realizar una gestión adecuada de los residuos generados.
– Cuantificación de productos de PCR
Como se ha mencionado anteriormente, la última clasificación de la ELN 2017 ha incluido el valor de la ratio “alelo-ITD/alelo-WT” de FLT3‐ITD. Es por ello necesario determinar el valor de expresión de ambos alelos. Los métodos clásicos de retrotranscripción del ARN, seguido de PCR a tiempo real no son aplicables en este caso, dado que las variantes son diferentes en cada paciente. Por ello se ha desarrollado un método cuantitativo basado en PCR a punto final. Se emplean cebadores que flanquean la región ITD (uno de ellos marcado con un fluoróforo), se amplifican, y luego se efectúa electroforesis capilar (EC) de los amplicones en un secuenciador automático del método Sanger. Este secuenciador separa por EC los amplicones procedentes de cada uno de los alelos, ya que son de diferente tamaño debido a la presencia de las ITDs y, por tanto, migran a velocidades distintas. Según el protocolo de Murphy y colaboradores, el alelo normal presenta un tamaño de amplificación de 328 pares de bases (Murphy et al., 2003). El secuenciador permite cuantificar el área bajo los picos de fluorescencia de ambos alelos, lo que permite calcular la ratio entre ambos. Aunque este método analítico resulta algo más caro que los métodos de PCR convencional por el empleo de reactivos que permiten el marcaje con fluorescencia y de un secuenciador Sanger, es indudable su ventaja frente a la PCR convencional por proporcionar la posibilidad de cuantificar la ratio según marcan las guías clínicas; además, es un protocolo relativamente rápido, con una sensibilidad estimada del 5% de alelo ITD (Daver et al., 2019) (Tabla 2).
| TÉCNICA | MUTACIONES | CUANTITATIVA | SENSIBILIDAD (%) |
TIEMPO DE RESPUESTA (días) |
COSTE |
| Basadas en PCR | dirigidas | no | 1% | 3 | reducido |
| PCR con marcación fluorescente | dirigidas | si | 5% | 3 | reducido |
| NGS | nuevas | por determinar | 1-5% | 15-20 | elevado |
Tabla 2. Comparativa de las metodologías actualmente en uso para el genotipado de FLT3. Los datos de sensibilidad son tomados de Daver et al. (Daver et al., 2019). Los datos de tiempo de respuesta y coste son tomados de nuestra propia experiencia.
PCR: Polymerase Chain Reaction; NGS: Next Generation Sequencing.
2. Detección de las variantes FLT3-TKD
Los métodos clásicos de detección de estas variantes se basan en la amplificación por PCR empleando cebadores específicos que flanquean la región donde se dan las variantes FLT3-TKD (exón 20), seguido del método elegido para detectar la variante.
– PCR con análisis de polimorfismos de longitud de los fragmentos de restricción (Restriction Fragment Length Polymorphism, RFLP)
Las mutaciones que afectan a los aminoácidos D835 e I836 localizadas en el TKD2 se pueden detectar por digestión enzimática con EcoRV del amplicón de PCR.
El fundamento de esta técnica reside en el hecho de que la presencia de la variante provoca la desaparición de la diana de esta enzima de restricción, de modo que el patrón de los fragmentos de ADN en un gel de agarosa o por electroforesis capilar permite determinar la presencia/ausencia de la variante. El protocolo descrito por Yamamoto y colaboradores produce un amplicón de 114 pares de bases. En el alelo normal (wild type, WT), la presencia de la diana de esta enzima de restricción permite la digestión del fragmento en 68 y 46 pares de bases, respectivamente (Yamamoto et al., 2001). En caso de que presente alelos variantes que afecten a los aminoácidos D835 o I836, la diana de digestión con EcoRV desaparece por lo que no hay digestión en dos fragmentos y el tamaño del fragmento es el de la PCR, 114 pares de bases. A continuación, se identifica la variante concreta mediante secuenciación Sanger.
Este procedimiento es un buen método de cribado, que evita el tener que realizar directamente secuenciación Sanger a todas las muestras a analizar, lo que abarata y agiliza el análisis de un elevado número de muestras. Además de ser económico, rápido, y fácil de interpretar, es posible procesar un alto número de muestras simultáneamente, y el equipamiento que precisa está disponible en cualquier laboratorio de Biología Molecular.
– Análisis de alta resolución de fusión (HRM)
Un método alternativo de detección de las variantes en los aminoácidos D835 e I836 es la High Resolution Melt (HRM) o análisis de alta resolución de fusión, que se basa en el estudio y comparación de las curvas de fusión de amplicones que incluyen estas variantes. Tras la amplificación por PCR del exón 20, se estudia el perfil de melting de los amplicones. Un perfil diferente al del control wild type sería indicativo de la presencia de la variante, que tendría que ser confirmada por secuenciación Sanger (Fuster et al., 2009; Liu et al., 2014). Al igual que el anterior, este procedimiento es un buen método de cribado, que abarata y agiliza el análisis de un elevado número de muestras. Sin embargo, esta ventaja puede convertirse en ocasiones en una limitación, ya que el sistema es dependiente de un número mínimo de muestras que deben ser analizadas simultáneamente, de modo que cuente con la potencia necesaria para detectar los perfiles anómalos; además, requiere una plataforma específica que no forma parte del equipamiento básico de un laboratorio de Biología Molecular.
En Abril de 2017 la FDA aprobó el uso de Leukostrat CDx FLT3 Mutation Assay, una prueba basada en este concepto que analiza simultáneamente la presencia de ITDs y variantes en los codones D835 e I836. Esta prueba, comercializada por Ia compañía de servicios diagnósticos Invivoscribe, está aprobada como técnica diagnóstica en el contexto del tratamiento con Rydapt (Midostaurina) en combinación con quimioterapia (Administration, 2017). La compañía declara una sensibilidad del ensayo de una célula portadora de ITD entre 10.000 células, siempre que se ensaye sobre una cantidad mínima de ADN procedente de 100.000 células, esto es, un mínimo de 660 ng (Levis et al., 2018). Además de este kit comercial, existen otros “métodos promesa” que permiten el análisis simultáneo de todas las variantes de secuencia de FLT3, como describimos a continuación.
3. Secuenciación de próxima generación (NGS)
En la última década, la aparición de la secuenciación masiva paralela (Next Generation Sequencing, NGS) ha supuesto una revolución en los laboratorios de diagnóstico genético, facilitando la transición al diagnóstico genómico, ya que esta tecnología permite el análisis de un buen número de marcadores en una única prueba. En concreto, en la mayoría de las herramientas NGS desarrolladas para el diagnóstico de la LMA se incluye el análisis del gen FLT3, con raras excepciones (Alonso et al., 2018). Sin embargo, los resultados que produce esta tecnología han de ser interpretados con precaución, ya que las características peculiares de FLT3-ITD complican su detección mediante NGS.
Por un lado, existe un reto de tipo químico. Es importante asegurar que en la preparación de librerías se captura el alelo variante. La longitud variable de la ITD hace que sea difícil de incorporar a las librerías en todos los casos; esto sucede tanto en los sistemas de captura por hibridación como en los sistemas de amplificación por PCR: si la ITD excede el tamaño medio de la librería, es probable que no se vea representada en la misma. Además, incluso en caso de que se llegue a capturar una ITD tan larga en la librería, la longitud máxima de lectura de los secuenciadores NGS actuales puede ser limitante para leer la ITD en toda su extensión, lo que provocaría que se perdiera la capacidad de determinar cuál es la longitud exacta de esa ITD.
Por otro lado, hay retos de tipo informático. Una vez secuenciada la librería, los archivos que generan las diferentes plataformas de secuenciación se pre-procesan, se alinean contra el genoma, y una vez alineados, se hace la detección de variantes. Los métodos tradicionales de alineamiento precisan una secuencia de bases que coincidan con el genoma de referencia tanto al principio de la secuencia como al final. Si no se cumple ese criterio, el alineador las descarta como secuencias basura. Por lo tanto, en el caso de las ITDs largas, aunque hayan sido incorporadas a la librería, cabe la posibilidad de que se pierdan en el proceso de alineamiento. Los nuevos métodos de alineamiento permiten realizar un ensamblado de las lecturas de novo, esto es, independiente del genoma de referencia, con lo que aumentan las posibilidades de detección de las ITDs que hayan sido incorporadas a la librería.
También es posible perder las secuencias portadoras de la inserción si el alineador sólo ha conseguido alinear las lecturas leídas en un único sentido. Cuando esto sucede, el algoritmo de identificación de variantes (variant caller) cataloga la inserción como “strand bias” (sesgo de hebra). Esta característica es un tipo de error relativamente frecuente debido a problemas durante la secuenciación, por lo que los criterios de calidad suelen recomendar eliminar estos tipos de variantes. En los pipelines informáticos automatizados esto resulta en la ausencia de estas inserciones con “strand bias” en la lista final de variantes.
En nuestra experiencia, el análisis de las ITDs con NGS en la mayoría de los casos es concordante con las técnicas de genotipado clásicas; sin embargo, otras veces hemos encontrado resultados discordantes. Por ejemplo, en casos donde las técnicas clásicas detectaban varios clones con distintos tamaños de ITDs, la secuenciación por NGS sólo identificaba alguno de ellos. Hemos observado una tendencia a que la NGS pierda las ITDs de mayor tamaño, hecho que ha sido insinuado recientemente en la literatura (Schranz et al., 2018).
En cualquier caso, aunque se llegaran a solucionar los retos químicos e informáticos arriba descritos, persiste la limitación antes mencionada procedente de las lecturas cortas de las plataformas de secuenciación de las que disponemos actualmente en los laboratorios de genética. A modo de ejemplo, mencionamos aquí las limitaciones declaradas por dos empresas que proporcionan herramientas NGS (tanto los kits para fabricar las librerías, como los algoritmos informáticos necesarios para analizar los resultados de secuenciación de las mismas). Sophia Genetics (Suiza) especifica que su panel mieloide (MYS, basado en captura por hibridación) es capaz de detectar ITDs de FLT3 de hasta 177 pares de bases de longitud (Sophia Genetics, 2018). En comunicación oral hemos sabido que han logrado detectar algún caso aislado de 244 pares de bases. Por otro lado, ArcherDX (Colorado, EE.UU.) utiliza una química diferente, basada en cebadores que hibridan en posiciones variables para amplificar las regiones de interés, y después analiza los resultados de secuenciación empleando un ensamblaje de novo, lo que en teoría les debería permitir la detección de ITDs de cualquier longitud. Sin embargo, la nota técnica de su panel mieloide muestra que detectan FLT3-ITD de hasta 180 pares de bases (Archer Dx, 2017). Estos dos ejemplos ilustran las limitaciones de la NGS para detectar FLT3-ITDs de cualquier tamaño, teniendo en cuenta que las ITDs pueden alcanzar longitudes cercanas a los 400 pares de bases. Este es otro motivo que apoya los datos incipientes de que en los en los pacientes con ITDs largas la NGS ofrezca falsos negativos (Schranz et al., 2018).
En el caso de las ITDs de longitud dentro de la capacidad de detección de la técnica, es teóricamente posible hacer el cálculo de la ratio entre el alelo mutado y el alelo wild type, empleando la variant allele frequency (VAF), al igual que se hace al cuantificar los productos de PCR por fluorescencia. Sin embargo, queda pendiente determinar cuál sería el punto de corte de VAF asociado a mal pronóstico en esta técnica, o si habría de ser un punto de corte dinámico, dependiente de la longitud de la ITD, ya que las distintas longitudes de ambos alelos podrían estar sesgando la eficiencia de la amplificación de los mismos durante el proceso de fabricación de las librerías. Efectivamente, la amplificación desigual de dos hebras de ADN de diferente tamaño en una misma reacción de PCR (como es el caso del alelo WT y del alelo portador de la ITD) es un fenómeno bien descrito que puede afectar al número de copias que son secuenciadas para cada alelo y, de hecho, se ha descrito para FLT3 (Polz y Cavanaugh, 1998; Schranz et al., 2018).
Por otro lado, es verdad que la gran capacidad de secuenciación de la NGS permite analizar toda la secuencia del gen en un único ensayo, por lo que es posible detectar tanto las variantes puntuales más frecuentes en los codones D835 e I836, como otras fuera del dominio TKD2 que han sido descritas más recientemente, como la mutación pN676K, encontrada en hasta el 6% de los casos de leucemias core-binding factor (Opatz et al., 2013; Patnaik, 2018).
Por último, hay que tener en cuenta que para ajustar costes, la técnica de NGS requiere agrupar un número mínimo de muestras para una carrera de secuenciación y que el proceso de generación de librerías, secuenciación y análisis requiere un tiempo que oscila entre 5 días y varias semanas por lo que se hace imposible dar un diagnóstico dentro de las 48-72 horas recomendadas por la ELN 2017, al menos en pacientes que vayan a recibir quimioterapia intensiva (Dohner et al., 2017).
CONCLUSIONES
La correcta determinación de las alteraciones genéticas del gen FLT3 es crucial desde el punto de vista clínico, tanto por su valor pronóstico asociado, como por la disponibilidad actual de terapias dirigidas a algunas de esas alteraciones. Las técnicas analíticas tradicionales ofrecen distintas ventajas e inconvenientes en cuanto al tiempo de respuesta y cuantificación de los alelos variantes. Entre ellas, destaca la utilidad de la cuantificación de fragmentos de PCR por fluorescencia, ya que permite establecer el valor de la ratio “FLT3-ITD/FLT3-WT” que señalan las guías. Sin embargo, esta técnica no analiza el conjunto de las alteraciones de FLT3, por lo que es necesario realizar otras pruebas genéticas complementarias.
La disponibilidad de la NGS ha hecho soñar con el concepto del just one test, una única prueba NGS que analiza la longitud completa del gen y que, por tanto, en teoría podría sustituir a todas las técnicas analíticas genéticas clásicas. Sin embargo, en base a ejemplos recientes que demuestran ciertas limitaciones técnicas de la NGS para detectar inserciones grandes, en este trabajo hacemos la reflexión de que conviene conocer a fondo esta tecnología y testar sus posibilidades de un modo sistemático antes de decidir eliminar otras pruebas basadas en tecnologías básicas de Biología Molecular. Además, por el momento, no se puede prescindir de las técnicas clásicas en la caracterización de las alteraciones genéticas de FLT3, ya que los resultados deben estar disponibles entre 48 y 72 horas para la aplicación de la terapia.
AGRADECIMIENTOS
Los autores quieren expresar su agradecimiento a todo el equipo de CIMA LAB Diagnostics (https://www.unav.edu/web/cimalab/cima-lab-diagnostics/equipo/genetica-de-enfermedades-hematologicas), y a todos los pacientes que participan en nuestros estudios. MFM agradece también financiación de la Asociación Española Contra el Cáncer (AECC), y del Instituto de Salud Carlos III (PI16/00159).
