INTRODUCCIÓN
En los últimos años se han producido avances muy importantes en el desarrollo de biomarcadores en el campo de la oncología que desempeñan un papel fundamental en la comprensión de los mecanismos moleculares y celulares que impulsan la iniciación, el mantenimiento y la progresión tumoral. Estos descubrimientos han impulsado el desarrollo de nuevas dianas farmacológicas y nuevas estrategias de tratamiento. De este modo, el manejo clínico de los pacientes oncológicos ha pasado en los últimos años de una estrategia de tratamiento empírico basada en el perfil clínico-patológico a una en la que se usa un algoritmo de tratamiento basado en biomarcadores que definen el perfil molecular del tumor. Esta aproximación ha permitido avanzar en los últimos años hacia una medicina personalizada de precisión que ha cambiado los paradigmas en oncología, permitiendo un diagnóstico más rápido y preciso y una mejor selección del tratamiento (Nalejska et al., 2014).
El estudio de las alteraciones epigenéticas se encuentra entre los biomarcadores que pueden tener un mayor impacto clínico en el campo de la oncología de precisión en los próximos años. Estas alteraciones epigenéticas se han identificado tradicionalmente en estudios realizados en genes individuales (Esteller et al., 2000). Sin embargo, la reciente implementación de nuevas tecnologías, como los sistemas de microarrays y la secuenciación masiva, está permitiendo el análisis de gran parte o la totalidad del epigenoma, lo cual producirá grandes cantidades de datos que mejorarán nuestra comprensión del desarrollo y la progresión del tumor y permitirá la identificación de un mayor número de biomarcadores epigenéticos con posibles aplicaciones clínicas (Diaz-Lagares et al., 2016b). Desde un punto de vista clínico es especialmente relevante el análisis de biomarcadores epigenéticos en biopsia líquida, ya que esto permitirá la detección del tumor y seguimiento de la enfermedad mediante análisis no invasivos, ofreciendo un mejor manejo de los pacientes con un seguimiento más personalizado y preciso (Mari-Alexandre et al., 2017).
CARACTERÍSTICAS DE LOS BIOMARCADORES
El Instituto Nacional del Cáncer (NCI) define los biomarcadores como moléculas biológicas que se encuentran en la sangre, otros fluidos corporales o tejidos, resultado de un proceso normal o anormal, o de una afección o enfermedad (Henry and Hayes, 2012). Cuando estas moléculas presentan utilidad en el contexto del cáncer reciben el nombre de biomarcadores tumorales y pueden ser empleados para la toma de decisiones clínicas. En este sentido, hay tres tipos clave de biomarcadores con aplicación clínica que son los biomarcadores de diagnóstico, de pronóstico y predictivos. Los biomarcadores de diagnóstico facilitan principalmente la identificación o clasificación de un tipo o subtipo concreto de cáncer. Mientras que, los de pronóstico ayudan a determinar el riesgo de recaída o progresión de la enfermedad después del tratamiento, de manera que los pacientes con alto riesgo pueden ser seleccionados para una terapia adyuvante que pueda prevenir la recurrencia de la enfermedad. Por otro lado, los biomarcadores predictivos permiten clasificar a los pacientes en función de la probabilidad de respuesta a ciertas terapias permitiendo así seleccionar la terapia más adecuada para cada paciente (Roychowdhury y Chinnaiyan, 2016).
Los biomarcadores tumorales empleados en la clínica deben de presentar idealmente varias de las siguientes características: (1) originarse por parte del tejido maligno o premaligno de manera específica y temprana en la progresión de la enfermedad; (2) producirse a niveles detectables en todos los pacientes con un tumor concreto; (3) producirse en un órgano o tejido de manera específica; (4) estar presente en fluidos biológicos obtenidos de manera no invasiva o en tejidos de fácil acceso; (5) correlacionarse cuantitativamente con el volumen del tumor, con su comportamiento biológico o con la progresión de la enfermedad; (6) presentar una vida media relativamente corta, reflejando así cambios temporales en la carga tumoral y en la respuesta a la terapia; y (7) disponer de una metodología estandarizada, reproducible, objetiva, cuantitativa y validada para su análisis (Bigbee, 2003).
LA MAQUINARIA EPIGENÉTICA: ENFOCÁNDONOS EN LA METILACIÓN DEL ADN Y ARN NO CODIFICANTE
La epigenética se refiere a cambios hereditarios en la actividad y expresión de los genes que se producen sin alterar la secuencia del ADN (Berger et al., 2009). Este mecanismo juega un papel importante regulando la expresión génica de muchos procesos biológicos que tienen lugar a lo largo de la vida de un individuo e ilustran la razón por la cual un organismo produce muchos tipos de células diferentes durante su desarrollo, a pesar del hecho de que la mayoría de las células en un organismo multicelular comparten la misma información genética (de Mello et al., 2014). La maquinaria epigenética presenta varios niveles de regulación: metilación del ADN, modificaciones de histonas, posicionamiento del nucleosoma y ARN no codificante (ncRNA) como los microARNs y los ARN largos no codificantes (lncRNAs) (Rodriguez-Paredes and Esteller, 2011). Entre estos mecanismos epigenéticos, la metilación del ADN y los ncRNAs (Figura 1) son dos de las modificaciones epigenéticas más ampliamente estudiadas (Esteller, 2008, Esteller, 2011).
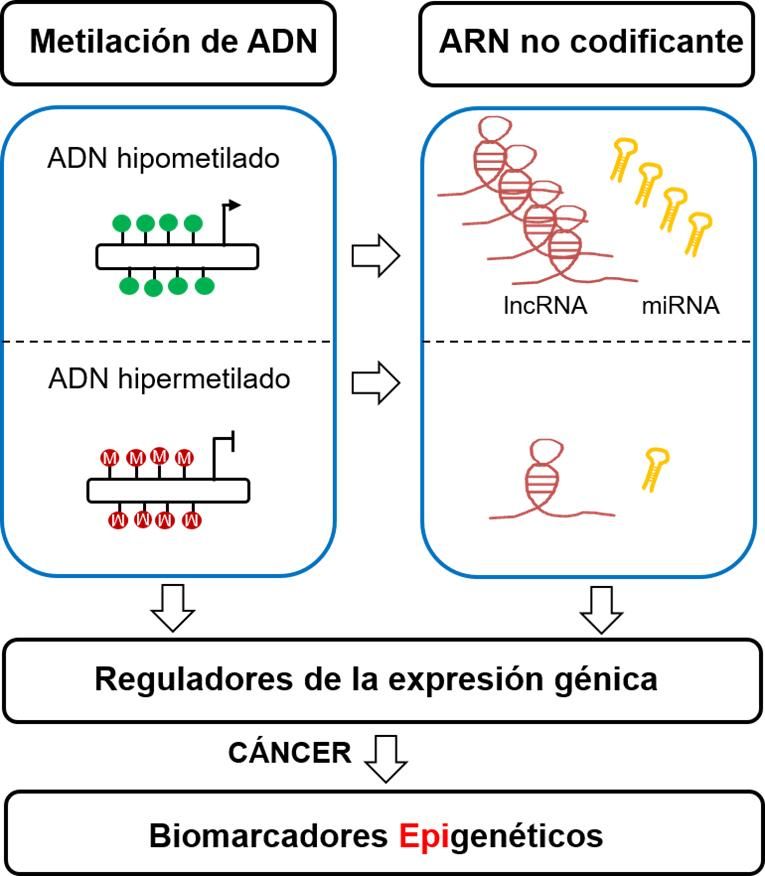
La metilación del ADN generalmente ocurre en ciertas áreas del genoma con una alta concentración de dinucleótidos CpG llamadas islas CpG (CGI) que conducen al silenciamiento tanto de los genes codificantes como de los genes no codificantes (Lujambio et al., 2007). Por otra parte, la metilación del ADN se produce también en otras regiones genómicas diferentes para mantener la conformación y la integridad de los cromosomas, así como para evitar el daño potencial de los elementos genéticos móviles (Herceg y Vaissiere, 2011). Además, la metilación del ADN puede afectar no solo a regiones intragénicas sino también a regiones intergénicas del genoma aumentando el nivel de complejidad de la interacción entre la metilación del ADN y la regulación de la expresión génica (Sandoval et al., 2011). Este mecanismo de metilación del ADN está controlado enzimáticamente por las enzimas ADN metiltransferasa (Dnmt), dentro de las cuales la Dnmt3A y 3B son esenciales para la metilación de novo, mientras que, la Dnmt1 mantiene los patrones de metilación durante la división celular (Gowher y Jeltsch, 2002; Jaenisch y Bird, 2003).
Por otro lado, también se ha demostrado que los ncRNAs juegan un papel importante en el control de la expresión génica (Esteller, 2011). Estudios recientes de todo el genoma han demostrado que el genoma humano produce diferentes tipos de ncRNAs reguladores (Taft et al., 2010), siendo los microARNs (miRNA) el tipo más ampliamente estudiado. Los miRNAs (de 18-25 nucleótidos) son moléculas monocatenarias que se unen a regiones específicas de ARN mensajero (ARNm) diana y median el silenciamiento génico postranscripcional al bloquear la transcripción o degradar el ARNm (Lee y Calin, 2011). A través de este mecanismo, los miRNAs están implicados en procesos importantes que incluyen el desarrollo, la proliferación, la diferenciación celular, la regulación del ciclo celular y la apoptosis (Esteller, 2011; Garzon et al., 2009).
El número de ncRNAs identificados en los últimos años está aumentando rápidamente. En concreto, se ha descrito recientemente que los lncRNAs constituyen la gran mayoría del transcriptoma no codificante con miles de estas especies de ARN identificadas hasta el momento (Hon et al., 2017). Aunque los lncRNAs carecen de potencial para codificar proteínas, pueden presentar algunas propiedades similares a los ARNm, como las estructuras génicas multiexónicas, poliadenilación, presencia de caperuza 5′ y transcripción por ARN polimerasa II (Derrien et al., 2012; Guttman et al., 2009). Los lncRNAs presentan una importante función de control de la expresión génica y se asocian con una gran variedad de funciones reguladoras, como el control del splicing y la regulación transcripcional (Kotake et al., 2011; Leveille et al., 2015). Aunque la mayoría de los lncRNAs aún no se han estudiado en detalle, algunas de estas moléculas se han caracterizado en cáncer, observándose que pueden actuar como oncogenes (HOTAIR y MALAT1) (Gupta et al., 2010; Gutschner et al., 2013) o como genes supresores de tumores (TP53TG1 y LED) (Diaz-Lagares et al., 2016a; Leveille et al., 2015).
LAS ALTERACIONES EPIGENÉTICAS COMO BIOMARCADORES EN CÁNCER
Las alteraciones epigenéticas se han descrito en diversas enfermedades, especialmente en el cáncer (Berdasco y Esteller, 2010), y han surgido en los últimos años como una herramienta con gran potencial clínico como biomarcadores (Figura 2). Entre los mecanismos epigenéticos, la metilación del ADN y los ncRNAs destacan como las alteraciones epigenéticas más estudiadas con utilidad clínica en cáncer, debido en parte a que poseen la gran mayoría de las características comentadas anteriormente que idealmente deben de cumplir los biomarcadores tumorales. Como uno de los primeros ejemplos de biomarcador con utilidad clínica destaca la metilación del gen reparador del ADN O-6-metilguanina-ADN metiltransferasa (MGMT) en muestras tumorales de pacientes con cáncer de glioblastoma, que permite establecer la respuesta clínica a la terapia con agentes alquilantes en este tipo de tumor (Esteller et al., 2000). Este biomarcador abrió el camino a la farmacoepigenética y se utiliza actualmente en la clínica, desempeñando un papel central como biomarcador en la clasificación de los gliomas (OMS 2016) y en las decisiones de tratamiento de este tipo de tumor (Gusyatiner y Hegi, 2017). Además de evaluar la respuesta a la terapia, los biomarcadores epigenéticos han mostrado ser útiles en el diagnóstico y clasificación de los tumores. Un ejemplo reciente es el desarrollo de una prueba epigenética denominada EPICUP basada en la detección de metilación de ADN en muestras de tejido tumoral con utilidad para identificar los tumores de origen desconocido o cancer of unknown primary (CUP). Esta prueba epigenética, que está comercializada por la empresa Ferrer, permite guiar mejor la terapia en función del origen del tumor mejorando la evolución de los pacientes (Moran et al., 2016) y representa un claro ejemplo de cómo la epigenética puede ser incorporada a la clínica. Resalta además que el análisis de la metilación de ADN empleada en la prueba EPICUP mostró una mayor robustez que otras pruebas diagnósticas empleadas hasta el momento para la detección de este tipo de tumores. Por otro lado, la metilación de ADN también ha mostrado ser útil como marcador pronóstico en diferentes tipos de tumores, como el cáncer de pulmón. Así, recientemente se ha observado que el estado de metilación del promotor del gen TMPRSS4 es un biomarcador con capacidad para predecir el pronóstico en los pacientes con estadios tempranos de cáncer de pulmón, lo cual podría contribuir a seleccionar a los pacientes con riesgo de recaída en etapas tempranas de la enfermedad (Villalba et al., 2016).
Aunque el estudio de las marcas de metilación del ADN tradicionalmente se ha centrado en los genes codificantes de proteínas, se ha demostrado recientemente que también existe una interacción entre este mecanismo epigenético y los genes que dan lugar a ncRNAs (Lujambio et al., 2007; Diaz-Lagares et al., 2016a). Este nuevo punto de vista se puede abordar mejor ahora con el desarrollo actual de las herramientas epigenómicas que pueden conducir a identificar nuevos ncRNAs alterados epigenéticamente como biomarcadores. Así, hay varios estudios epigenómicos que han encontrado una asociación entre el silenciamiento de la expresión de los ncRNAs y la metilación del ADN. Además de los miRNAs, otros tipos de ncRNAs han mostrado presentar alteraciones epigenéticas. Recientemente, un estudio epigenómico de metilación del ADN mediante sistemas de microarrays de metilación reveló la inactivación epigenética en cáncer de un lncRNA inducido por p53 denominado TP53TG1. En concreto, la hipermetilación de TP53TG1 se correlacionó con la resistencia a múltiples fármacos usados en la práctica clínica y también mostró asociación con un peor pronóstico para los pacientes en tumores gastrointestinales, indicando una posible utilidad como biomarcador epigenético en este tipo de tumores (Diaz-Lagares et al., 2016a). Otro estudio reciente con un enfoque diferente, basado en un análisis transcriptómico de lncRNAs, permitió identificar otro lncRNA inducido por p53 denominado LED (LncRNA activator of Enhancer Domains). En este trabajo se mostró que LED está regulado epigenéticamente mediante metilación de ADN y se encuentra hipermetilado en pacientes con diferentes tipos de leucemias, por lo que podría tener aplicaciones como biomarcador tumoral (Leveille et al., 2015). En definitiva, perfilar la metilación del ADN mediante estudios epigenómicos se ha convertido en una estrategia fundamental para comprender la influencia de la epigenética tanto en la biología como en situaciones de enfermedad. Este tipo de estrategias representa una gran promesa y podría guiar diagnósticos y terapias más precisas asociadas con mejores resultados para los pacientes con cáncer.
De forma similar a la metilación de ADN, el análisis de los niveles de expresión de ncRNAs (especialmente miRNAs y lncRNAs) en muestras tumorales presenta una gran relevancia como posible biomarcador tumoral. Los primeros trabajos en evidenciar la utilidad de los patrones de expresión de miRNAs como herramientas para clasificar los tumores mostraron que estos perfiles no sólo permitían distinguir el origen del tumor sino que también daban información sobre su grado de diferenciación y conseguían clasificar a los tejidos tumorales poco diferenciados (Lu et al., 2005). Debido a su elevada capacidad para diferenciar los subtipos de cáncer, varias compañías biotecnológicas han desarrollado productos de diagnóstico basados en niveles de expresión de miRNAs. De este modo, mediante el uso de 8 miRNAs, mi-LUNG (Rosetta Genomics Ltd.) es capaz de identificar los diferentes subtipos de cáncer de pulmón con una elevada sensibilidad (94%) y especificidad (98%) (Gilad et al., 2012). Además de su utilidad diagnóstica, diversos trabajos han sugerido que la detección de miRNAs en muestras tumorales puede presentar también utilidad pronóstica en varios tipos de cáncer, como es el caso de miR-224 y miR-200a en cáncer de colon (Ling et al., 2016; Pichler et al., 2014), y miR-155 en linfomas (Iqbal et al., 2015), entre otros. Por otro lado, hay también un creciente interés en identificar miRNAs que se encuentren asociados a la terapia antitumoral. Este es el caso por ejemplo de miR-30e, cuya expresión ha sido recientemente asociada con un aumento de la sensibilidad a la terapia en cáncer de pulmón (Ning et al., 2017).
El hecho de que los lncRNAs estén altamente desregulados en varios tipos de tumores y presenten un alto grado de especificidad de tejido y enfermedad los convierte también en candidatos ideales para su uso en la clínica. En este sentido, la aplicación más conocida de estas moléculas hasta el momento ocurre en el diagnóstico del cáncer de próstata, donde el PCA3 (Prostate Cancer Antigen 3, también conocido como DD3) se comporta como un lncRNA específico de la próstata que se sobreexpresa múltiples veces en la mayoría de los pacientes con tumores de próstata, en comparación con el tejido prostático de las alteraciones benignas, y es indetectable en otros tipos de tumores (Hessels et al., 2003). Además del diagnóstico, los lncRNAs, como MALAT-1 por ejemplo, han mostrado también potencial como biomarcadores para el pronóstico y para predecir la recurrencia en varios tipos de cáncer (Xu et al., 2015; Wang et al., 2017). Sin embargo, ninguno de estos lncRNAs ha alcanzado todavía un uso clínico generalizado. Este hecho puede ser debido en parte al limitado número de lncRNAs estudiados hasta el momento, la falta de cohortes grandes de pacientes y el uso de métodos de detección con baja sensibilidad para este tipo de moléculas que, habitualmente, presentan rangos de expresión muy inferiores a los ARNms. Por tanto, serían necesarios estudios clínicos controlados con un elevado número de pacientes para poder implementar su uso en la clínica. Además, la combinación de dos o más lncRNAs o de estas moléculas con otro tipo de biomarcadores podría aumentar la sensibilidad y la especificidad de los biomarcadores existentes para la predicción de la respuesta terapéutica y así poder manejar la enfermedad antes de que se produzca su recurrencia (Chandra Gupta y Nandan Tripathi, 2017).
BIOMARCADORES EPIGENÉTICOS Y BIOPSIA LÍQUIDA
En los últimos años la biopsia líquida ha emergido como una herramienta clínica de gran importancia en el campo de la oncología. Esta nueva técnica consiste en analizar el material circulante presente en los fluidos biológicos que proviene de los tumores. La biopsia líquida incorpora grandes ventajas a la práctica clínica, ya que permite una detección no invasiva de los tumores, monitorizar mejor la respuesta al tratamiento, cuantificar la enfermedad mínima residual y evaluar la aparición de resistencia a la terapia (Mari-Alexandre et al., 2017). En la actualidad, este tipo de análisis se realiza mayoritariamente mediante el estudio del tumor tras la obtención de biopsias del tejido tumoral. Sin embargo, este tipo de muestra presenta algunas limitaciones que la biopsia líquida podría contribuir a solventar, ya que los fluidos biológicos son muestras más accesibles y su obtención es usualmente menos invasiva y más segura. La biopsia líquida presenta además la posibilidad de realizar múltiples extracciones longitudinales que permiten seguir la evolución del tumor para conocer los mecanismos de resistencia secundaria al tratamiento y la heterogeneidad tumoral en pacientes con múltiples tumores distribuidos por el cuerpo y a los que no se puede acceder para obtener muestra de tejido tumoral (Siravegna et al., 2017). Aunque en la biopsia líquida se analiza principalmente la sangre, el material tumoral circulante también puede encontrase en muchos otros fluidos como la orina (Deras et al., 2008), saliva y otros fluidos respiratorios (Diaz-Lagares et al., 2016b), líquido cefalorraquídeo (De Mattos-Arruda et al., 2015) y heces (Imperiale et al., 2014).
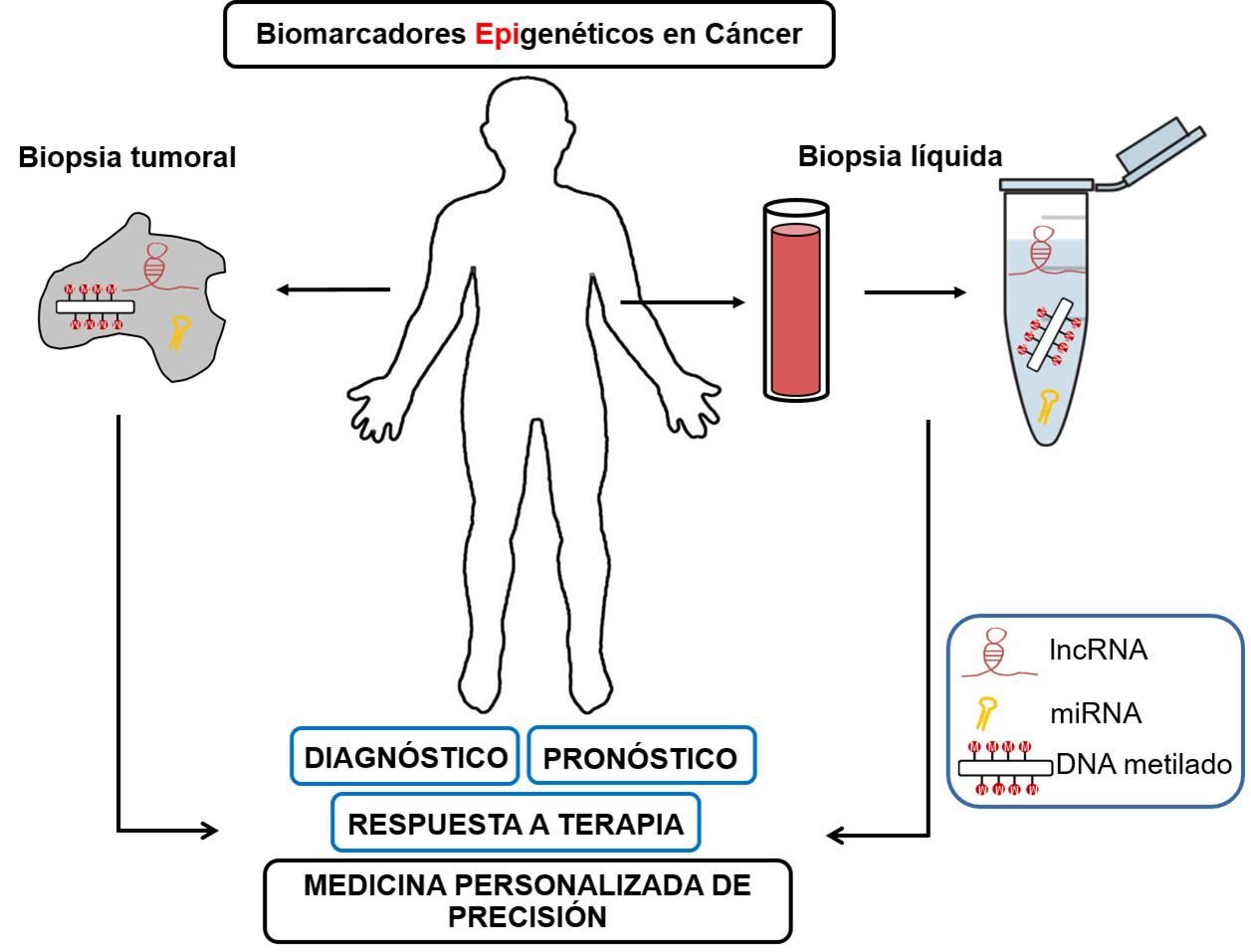
Entre el material tumoral circulante que puede detectarse en los fluidos biológicos se encuentran importantes componentes de la maquinaria epigenética, como la metilación del ADN y los ncRNAs (Figura 2). Así, varios estudios han obtenido una buena correlación entre las marcas epigenéticas en tejidos tumorales y las muestras de sangre en pacientes con diferentes tipos de tumores, indicando que las alteraciones epigenéticas circulantes son un buen reflejo de las modificaciones epigenéticas del tumor (Kadam et al., 2012). En este sentido, recientemente hemos descrito una firma epigenética para el diagnóstico precoz de cáncer de pulmón identificada primero en muestras de tejido tumoral y posteriormente validada en muestras de biopsia líquida (Diaz-Lagares et al., 2016b). Esta firma epigenética está basada en la metilación de 4 genes (BCAT1, CDO1, TRIM58, y ZNF177) que fueron identificados mediante un análisis epigenómico con sistemas de microarrays de metilación y validadas mediante métodos de fácil implementación clínica, como la pirosecuenciación, en muestras de fluidos respiratorios (aspirados bronquiales, lavados bronquioalveolares y esputos) de diferentes cohortes independientes. Esta firma epigenética mostró una elevada eficacia diagnóstica para detectar el cáncer de pulmón en sus fases iniciales (estadio I) con áreas bajo la curva ROC (AUC) en torno a 0.90. Estos resultados superaron las capacidades diagnósticas mostradas en este tipo de fluidos por la citología convencional para el diagnóstico de cáncer de pulmón. Este modelo epigenético en combinación con los protocolos diagnósticos actuales podría mejorar el diagnóstico temprano y la evolución de los pacientes con cáncer de pulmón.
Hasta el momento hay un considerable número de biomarcadores basados en metilación de ADN que han sido estudiados en sangre en relación con distintos tipos de tumores. Sin embargo, solo uno de estos biomarcadores ha sido a día de hoy aprobado por la Administración de Medicamentos y Alimentos de Estados Unidos (FDA). Este es el caso del test Epi proColon (Epigenomics), que representa el primer test aprobado por la FDA (2016) en sangre para el cribado de cáncer colorrectal para pacientes que no desean o no puedan someterse a exámenes de detección con los métodos recomendados habitualmente, como la colonoscopia, la sigmoidoscopia y las pruebas de sangre oculta en heces de alta sensibilidad u otros métodos de detección recomendados. La prueba Epi proColon ha sido evaluada en varios ensayos clínicos (PRESEPT NCT00855348; NCT01580540;) mostrando sensibilidades y especificidades en torno al 70% y 90%, respectivamente (deVos et al., 2009; Johnson et al., 2014). Esta prueba epigenética consiste en el análisis de la metilación en sangre del gen Septin 9 (SEPT9) mediante PCR en tiempo real y puede detectarse en el ADN tumoral circulante que se ha liberado tanto desde el tejido tumoral como desde lesiones precancerosas (adenomas), convirtiéndolo en un biomarcador diferencial para la detección temprana de cáncer de colorrectal (deVos et al., 2009).
Además del estudio de genes individuales, el análisis de paneles de múltiples genes puede contribuir a aumentar la sensibilidad y especificidad de firmas de metilación de ADN, como ocurre con el test Cologuard (Exact Sciences), que es otra prueba de cribado de cáncer colorrectal pero basada en este caso en el análisis de muestras de heces. Esta prueba, que representa el primer test diagnóstico basado en metilación aprobado por la FDA en el 2014, se basa en la combinación del análisis de metilación de los genes BMP3 y NDRG4 con la detección de mutaciones en KRAS y el análisis de hemoglobina oculta en heces (Imperiale et al., 2014). Sin embargo, el estudio de DNA fecal presenta algunas limitaciones para el cribado de cáncer colorrectal que podrían retrasar su implementación en la clínica, ya que su coste/eficacia parece ser menor que las pruebas actuales de sangre oculta en heces y colonoscopia (Song et al., 2004; Skally et al., 2013).
De manera similar a la metilación de ADN, los niveles de expresión de ncRNAs también se encuentran alterados en muestras de biopsia líquida de pacientes con cáncer. Así, en 2008 se demostró por primera vez la presencia de microARNs en fluidos biológicos (Lawrie et al., 2008), mostrando que los microRNA circulantes eran claramente detectables en muestras de suero y que los niveles más altos de microARNs específicos se asociaban con el diagnóstico y el pronóstico en pacientes con linfoma B difuso de células grandes (LBDCG), sugiriendo que estas moléculas tenían potencial como biomarcadores no invasivos con utilidad clínica. Este tipo de estudios podrían realizarse en una amplia variedad de fluidos biológicos debido a que se han detectado miRNAs en al menos 12 tipos de fluidos corporales, como plasma, orina, saliva, entre otros (Weber et al., 2010). Este tipo de moléculas circulantes cuenta con una notable estabilidad y relativamente fácil manejo, lo que les convierte en buenos candidatos para ser implementados en los laboratorios clínicos (Ono et al., 2015). Además de los miRNAs, los lncRNA circulantes también han demostrado ser útiles como biomarcadores tumorales. En plasma por ejemplo la detección del lncRNA GAS5 se ha sugerido como un biomarcador para el cribado del cáncer de pulmón de células no pequeñas (NSCLC) y la monitorización del paciente después del tratamiento quirúrgico (Tan et al., 2017). Sin embargo, la prueba en biopsia líquida basada en un lncRNA con más repercusión clínica hasta el momento la representa el ensayo en orina PROGENSA PCA3, aprobado por la FDA en 2016, basado en la detección del lncRNA PCA3 en pacientes con cáncer de próstata. PCA3 es un lncRNA específico de próstata implicado en el control de la supervivencia de las células de cáncer de próstata que se libera en la orina después del masaje prostático de los pacientes, ayudando a determinar si existe la necesidad de repetir la biopsia de próstata en hombres con una biopsia negativa previa (de la Taille, 2007; Lemos et al., 2016). La detección de PCA 3 en orina se ha utilizado como un biomarcador diagnóstico del cáncer de próstata con una sensibilidad y especificidad del 82% y 76%, respectivamente (Tinzl et al., 2004), mostrando ser incluso superior a pruebas usadas actualmente como el antígeno prostático específico (PSA) en suero (Ouyang et al., 2009).
CONCLUSIONES
Los biomarcadores epigenéticos son un área prometedora de investigación que presenta el potencial clínico de proporcionar una gran cantidad de información sobre el estado de la enfermedad tumoral. Diferentes etapas y tipos de cáncer reflejan una firma epigenética única, de manera que estas firmas pueden implementarse como biomarcadores específicos para establecer el tipo de tumor y ayudar a tener un diagnóstico y pronóstico más precisos, así como una mejor selección y manejo del tratamiento del cáncer. Es importante destacar que los marcadores epigenéticos pueden ayudar en la detección de tumores desde sus primeras etapas, lo que los convierte en una herramienta importante para el diagnóstico precoz y la detección de enfermedad mínima residual que podría incorporarse al conjunto actual de métodos de detección utilizados en la clínica. Aunque diferentes estudios han demostrado que los biomarcadores epigenéticos tienen relevancia clínica, para facilitar el uso generalizado de estos biomarcadores en la clínica se requiere de una mayor estandarización y validación. Además, es necesario avanzar también en la detección de estos biomarcadores en muestras de biopsia líquida, que permitirían un análisis no invasivo de los biomarcadores epigenéticos. Así, debido a su elevada precisión, especificidad y facilidad metodológica, las alteraciones epigenéticas tienen un gran potencial para convertirse en biomarcadores tumorales de rutina clínica, pudiendo llegar a implementarse paneles de biomarcadores epigenéticos no invasivos para el manejo clínico de los diferentes tipos de tumores tanto a nivel diagnóstico o pronóstico como para la selección y evaluación de la terapia más adecuada a cada paciente. Para alcanzar estos objetivos será también fundamental la ya iniciada implementación de las tecnologías ómicas al campo de la epigenética (epigenómica), con el uso de sistemas de microarrays y de secuenciación masiva, así como de tecnologías cada vez más sensibles, basadas por ejemplo en PCR digital, que permitirán detectar alteraciones epigenéticas que estén poco representadas en muestras no invasivas como los fluidos biológicos. De este modo, los biomarcadores epigenéticos, analizados tanto en muestras tumorales como en biopsia líquida, constituyen uno de los pilares importantes para alcanzar una oncología de precisión con enormes beneficios para los pacientes.
AGRADECIMIENTOS
La realización de este trabajo ha sido posible gracias a la financiación “Juan Rodés” (JR17/00016) recibida por ADL por parte del Instituto de Salud Carlos III (IISCIII).


