
El diagnóstico de las neoplasias hematológicas ha cambiado sustancialmente en las últimas décadas, pasando de la evaluación morfológica como único criterio, a la integración de los hallazgos clínicos, morfológicos, inmunofenotípicos, citogenéticos y moleculares, que son la base de la clasificación de la Organización Mundial de la Salud (OMS). Los nuevos datos genéticos han transformado nuestra comprensión de la fisiopatología de las neoplasias hematológicas, implicando nuevas vías biológicas y proporcionando una nueva herramienta para deconstruir la complejidad de dichas patologías. En los últimos años ha habido una explosión de nuevos datos moleculares gracias al uso de las técnicas de secuenciación masiva (Next-Generation Sequencing, NGS), tanto de genomas completos (WGS), exomas (WES), como paneles dirigidos al análisis de ciertos genes (Fernandez-Mercado, 2013; Dubois, 2016). La reciente Special Series del Journal of Clinical Oncology viene a reforzar esta visión de la importancia de la medicina genómica en oncología hematológica (Ebert, 2017).
Estamos presenciando de nuevo un momento crucial de cambio de paradigma en el diagnóstico onco-hematológico, con la incorporación del concepto de medicina de precisión (también llamada medicina personalizada). Ya vivimos una situación similar (de cambio profundo) hace tres décadas con la incorporación del análisis citogenético a nuestros laboratorios. Actualmente el cariotipo/FISH son esenciales para el diagnóstico, clasificación, estratificación pronóstica y orientación terapéutica en las hemopatías malignas. En este momento, el nuevo reto que tenemos por delante es la incorporación de la secuenciación masiva al diagnóstico integrado de las neoplasias hematológicas.
Es cierto que llevamos años realizando rutinariamente, además de técnicas citogenéticas convencionales (cariotipo y FISH), algunas pruebas moleculares que son necesarias tanto para el diagnóstico, como para la estratificación pronóstica en linfomas, mieloma múltiple, leucemia linfocítica crónica y otras neoplasias linfoides (Campo, 2011; Swerdlow, 2016; Rosenquist, 2016). Esto mismo sucede en patología mieloide, con el análisis de CEBPA, NPM1 y FLT3 en la leucemia mieloide aguda (LMA); aunque esta lista de pruebas moleculares podría alargarse en breve, ya que datos recientes apuntan a que hay mutaciones en otros genes que también podrían tener valor clínico en LMA (Papaemmanuil, 2016). En el caso de los síndromes mielodisplásicos (SMD), también se ha sugerido que el IPSS-R (Revised International Prognostic Scoring System, Greenberg, 2012) se podría mejorar mediante la incorporación de datos moleculares (Nazha, 2016). Como decimos, estos marcadores moleculares ya se están analizando con fines diagnósticos mediante técnicas de biología molecular clásica (secuenciación Sanger, ARMS-PCR, ASO-PCR o RT-qPCR), integrados con los datos de citomorfología, citometría y citogenética.
Sin embargo, la elevada cantidad de nuevos datos moleculares disponibles, junto con la accesibilidad de la NGS, ha abierto una nueva puerta a poder implementar los nuevos marcadores a los protocolos de los laboratorios diagnósticos de patología molecular, de modo que el diagnóstico genético que realizamos actualmente puede llegar a ser genómico. De hecho, diversas casas comerciales que incluyen secuenciación masiva en su cartera de productos están expandiendo su oferta de paneles NGS diseñados para analizar patologías específicas. Estos paneles dirigidos son capaces de analizar simultáneamente regiones de 20-60 genes, o incluso genes completos, seleccionados en base a la relevancia en la patología a estudiar. En términos económicos, la gran cantidad de información genómica que produce la tecnología de secuenciación masiva mediante paneles resultaría más cara y más laboriosa de obtener mediante secuenciación por el método de Sanger. Además, el análisis simultáneo de tan elevado número de genes permite reducir drásticamente (incluso a la mitad) los tiempos de respuesta, frente a los esquemas de análisis secuencial de genes que con frecuencia se aplican en los laboratorios moleculares (Figura 1). A las indudables ventajas en términos de tiempo y costes, se suma que esta tecnología posibilita la detección de clones emergentes que pueden ayudar en el seguimiento y pronóstico de la enfermedad, y detectar así un posible cambio de curso en su historia, quizá asociado a resistencia a tratamiento o a otros factores. Por último, disponer de una información molecular tan detallada permitirá la rápida identificación de pacientes que puedan beneficiarse de ensayos clínicos que contemplen dichas aberraciones moleculares en sus criterios de inclusión.
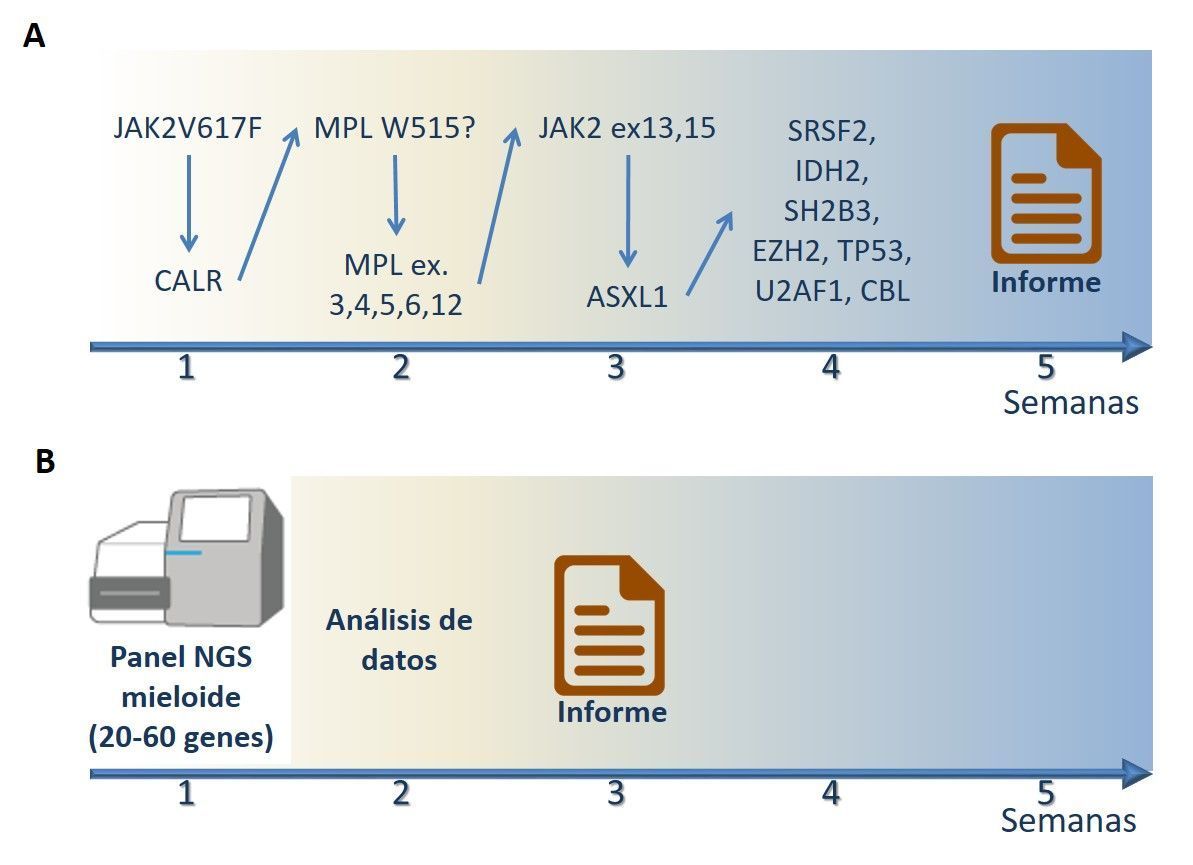
Sin embargo, hay algunos problemas que plantea la incorporación de la secuenciación masiva al diagnóstico, y que están retrasando la estandarización de su utilización. Por un lado, la complejidad de los datos que generan los secuenciadores requiere de personal altamente cualificado en biología computacional, lo que por el momento sólo es posible en laboratorios diagnósticos que tengan un fuerte compromiso con la investigación. Por otro lado, la escasez de guías en el ámbito internacional para el diagnóstico onco-hematológico provoca que los laboratorios que han empezado a emplear esta técnica tengan que hacer un esfuerzo grande de visión de conjunto de los resultados que van generando, para detectar errores recurrentes en la secuenciación, discernir las posibles implicaciones de hallazgos incidentales, desarrollar líneas de análisis de datos adaptadas a cada panel, y elaborar informes que sean de verdadera utilidad clínica. Finalmente, desde el punto de vista técnico, tenemos experiencia de que no todos los genes se analizan con igual precisión por esta técnica. Por ejemplo, con frecuencia las inserciones y deleciones largas escapan a los análisis informáticos estándar que se aplican sobre los datos de secuenciación; de modo similar, las regiones genómicas que están codificadas en sitios ricos en GC son difíciles de capturar en los pasos iniciales de preparación de la muestra antes de la secuenciación. Por estos motivos, prevemos un futuro próximo en el que probablemente la NGS no vaya a suplir a las técnicas moleculares ya instaladas en los laboratorios, sino que vaya a convivir con ellas, al menos durante unos años.
Además, las plataformas de secuenciación masiva generan tal cantidad de datos, que junto a las variantes de las que se conoce su relación con la enfermedad, se identifican otras de significado dudoso. Los laboratorios que incorporen la NGS al diagnóstico integrado de las neoplasias hematológicas se beneficiarán del trabajo coordinado con otros laboratorios que empleen las mismas técnicas, ya que juntos será más sencillo construir la masa crítica de datos de secuenciación masiva que permita interpretar el significado de las variantes genéticas de significado incierto (variants of uncertain significance, VUS). Por el momento, el recurso es acudir a bases de datos de variantes con datos de frecuencia poblacional para inferir si las VUS detectadas pueden guardar relación con la patología o no.
Por otro lado, es posible que esta tecnología no se extienda indiscriminadamente a todos los laboratorios de diagnóstico que en la actualidad realizan pruebas moleculares, sino que se restrinja a unos pocos laboratorios de referencia repartidos por el territorio nacional. Esto se podría deber, no sólo a la elevada inversión inicial en equipamiento y en la necesidad de disponer de personal altamente cualificado, sino también a que para poder garantizar buenos tiempos de respuesta y precios asequibles ha de haber un flujo de muestras suficiente.
Estamos presenciando un momento histórico, un nuevo punto de inflexión en el diagnóstico hematológico. La NGS ya no es sólo una tecnología de los grandes grupos de investigación, sino que se está implementando en el diagnóstico de rutina. Somos conscientes de que esto requerirá de intensa colaboración entre los especialistas en nuestro país en cuestiones como la estandarización de las técnicas (avalada por la SEHH), la formación de los profesionales o la acreditación de los procedimientos. Clínicos, anatomopatólogos, bioinformáticos y genetistas tenemos que trabajar más coordinados que nunca para que esta nueva tecnología de enorme potencial llegue a implementarse como nueva técnica diagnóstica, y sea de verdadero beneficio clínico.
Agradecimientos
Los autores quieren expresar su agradecimiento a todo el equipo de CIMA LAB Diagnostics. MFM agradece también financiación de la Asociación Española Contra el Cáncer (AECC), la Diputación Foral de Guipuzcoa (DFG15/011) y el Instituto de Salud Carlos III (PI16/00159).

