TECNOLOGÍAS DE SECUENCIACIÓN MASIVA: PRIMERAS GENERACIONES
Desde que en los años 70 se presentó el primer método de secuenciación desarrollado por Sanger hemos asistido a una revolución de las tecnologías de secuenciación genética, incentivada por el Proyecto Genoma Humano, culminado en el año 2001, que llevó a la secuenciación por vez primera del genoma completo (Roberts, 2001).
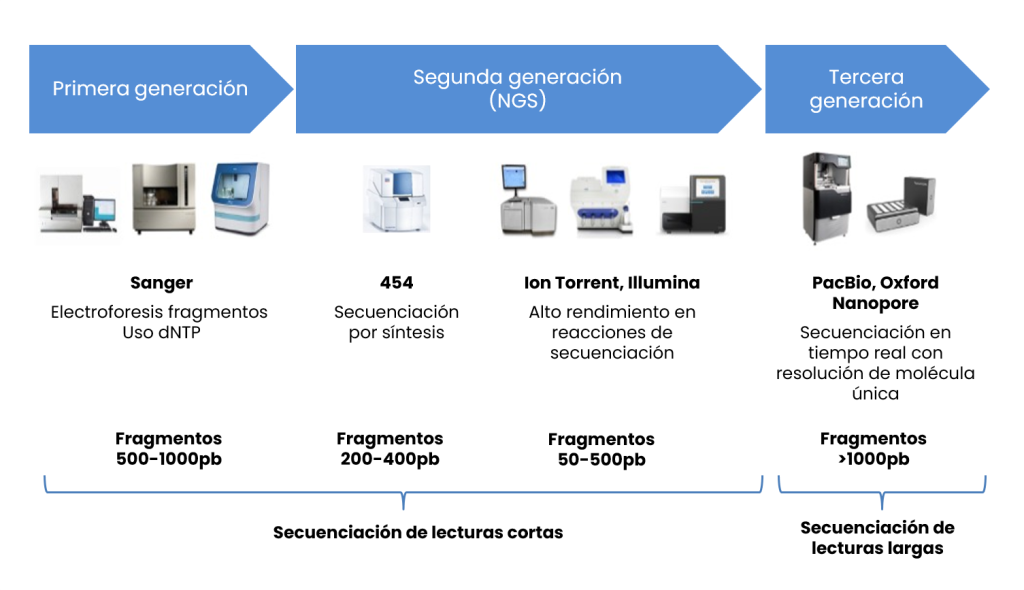
En 2005, la pirosecuenciación desarrollada por 454 Life Sciences, que tendría una trayectoria limitada, abrió el camino de las plataformas de secuenciación masiva denominadas “Next Generation Sequencing” (NGS) o de segunda generación, que permitían analizar múltiples muestras en paralelo.
Posteriormente, el desarrollo por compañías como Illumina e Ion Torrent de nuevas plataformas de NGS, globalizó la metodología pues hizo accesible equipamientos asequibles, capaces de secuenciar genomas completos en un tiempo y coste impensable a principios del s. XXI (Figura 1). Estas técnicas se basan en un proceso de segmentación del ADN por fragmentación o amplificación para su posterior lectura.
Finalmente, en 2012 surgió la secuenciación de “tercera generación”, liderada por Oxford Nanopore y PacBio, con tecnologías capaces de secuenciar el ADN o ARN en tiempo real sin necesidad de fragmentar ni amplificar el ADN (Figura 2).
Limitaciones de secuenciación de segunda generación
A pesar de que las técnicas de NGS son muy potentes para la detección de variantes puntuales y pequeñas indels, ya que son capaces de obtener de gran profundidad de lectura, el hecho de que se sustentan en lecturas de secuencias cortas hace que no sean tan robustas en la detección de variantes estructurales, la definición de grandes haplotipos, la secuenciación y correcto alineamiento en regiones repetitivas del genoma, o la caracterización de isoformas de ARN. Además, estos sistemas no identifican variaciones epigenéticas o epitranscriptómicas.
Estas limitaciones se ven resueltas con la llegada de las tecnologías de secuenciación de cuarta generación de lecturas largas, en especial la que emplea nanoporos, ya que, además, puede aportar nuevas perspectivas para resolver problemas genéticos más complejos.
SECUENCIACIÓN DE CUARTA GENERACIÓN CON NANOPOROS
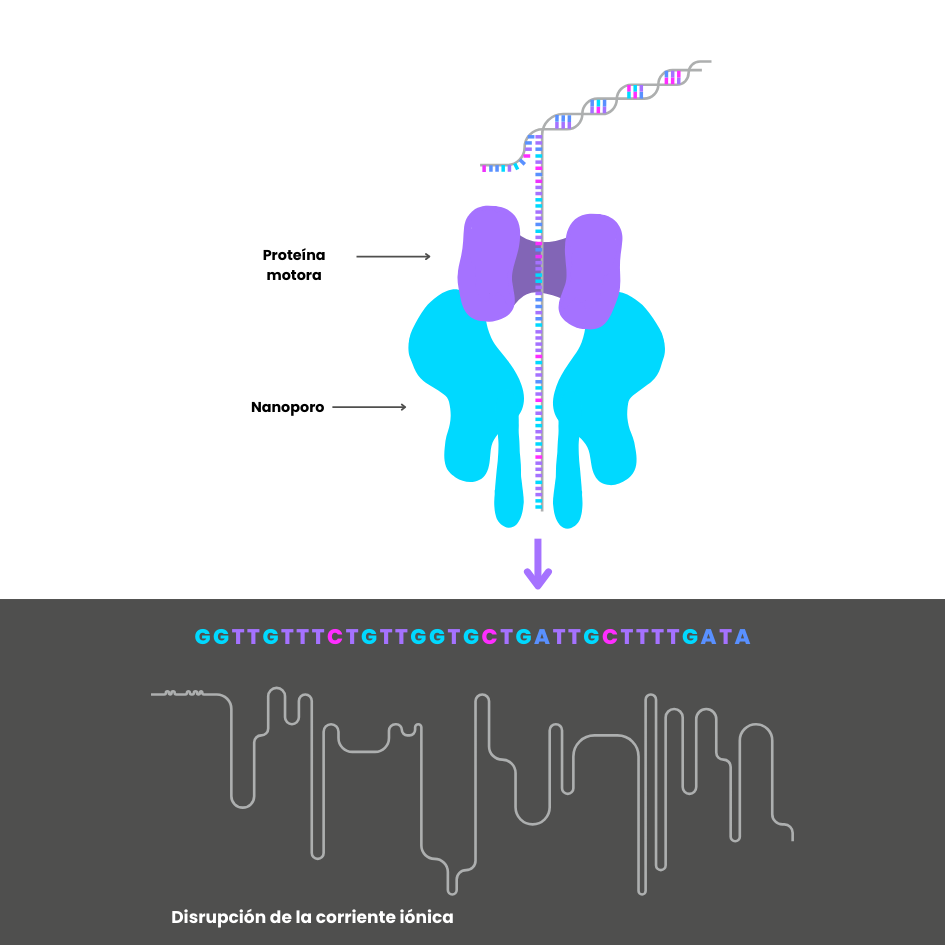
La secuenciación de cuarta generación mediante nanoporos, desarrollada por la empresa Oxford Nanopore Technologies, se basa en el cambio de potencial de corriente iónica que producen los nucleótidos de ADN o ARN al pasar por los nanoporos en la celda de carga o “flowcell” (Figura 3).
Existen diferentes dispositivos de secuenciación por nanoporos: MinION muy económico, con gran accesibilidad, pero de menor rendimiento, ya que solo tiene capacidad para secuenciar 5–10 Gb en 48 horas, y PromethION, con capacidad de secuenciar 2, 24 o 48 celdas de carga, con mayor rendimiento pues consigue 50-100 Gb en 72 horas en cada celda de carga.
La principal limitación de este sistema es su limitada cobertura, que es, en términos medios de 3x empleando secuenciación del genoma completo en MinION, o 30x empleando PromethION. No obstante, el poder realizar la secuenciación en tiempo real, permite un proceso de enriquecimiento denominado “adaptive sampling” que permite expulsar secuencias que no se alinean en la región seleccionada y aumentar la cobertura de dicha región, que se describirá con detalle más adelante.
Las principales ventajas que ofrece esta tecnología es que permite la obtención de lecturas extralargas de material genético (hasta 4 Mb). Además, el material genético se secuencia sin manipular y en tiempo real. Estas características permiten aplicaciones como la detección y caracterización completa de variantes estructurales, la definición de grandes haplotipos y diferenciación de alelos, el análisis epigenético de ADN o ARN, o el estudio de transcritos alternativos. En esta revisión, explicaremos brevemente estas posibilidades con la experiencia que hemos obtenido en nuestro grupo.
APLICACIONES DE LA SECUENCIACIÓN POR NANOPOROS
Estudio de variantes estructurales
Las variantes estructurales son alteraciones genéticas heterogéneas (deleciones, duplicaciones, inserciones, inversiones, translocaciones) que afectan a más de 50 pb, que son menos frecuentes que las variantes puntuales o pequeñas inserciones o deleciones, pero con mucha mayor carga patogénica (Stankiewicz et al., 2010).
Existen distintas técnicas que se han desarrollado para la detección de variantes estructurales, algunas de ellas que estudian el genoma completo (CGH array, SNP microarray, Cariotipo…) o dirigidas a regiones específicas (MLPA, FISH) (Tabla 1), pero todas tienen importantes limitaciones, como la incapacidad de definir la extensión exacta de la alteración, que el reciente sistema de mapeo óptico si consigue, aunque ninguno de estos sistemas alcanza resolución nucleotídica (Wan, 2017; Wan, 2014).
| Método | Translocaciones | Inversiones | CNV (>50 Kb) | INDELS (1-50 Kb) | Pequeñas VE (<1Kb) | Punto de corte |
| Genoma completo | ||||||
| Cariotipo | Sí (>3 Mb) | Sí (>3 Mb) | Sí (>3 Mb) | No | No | No |
| Array de CGH | No | No | Sí (>50 Mb) | No | No | No |
| SNP array | No | Sí | Sí | Sí | Sí (SNPs) | No |
| Paired-end sequencing | Sí | Sí | Sí | Sí | No | Si |
| Nanopore technology | Sí | Sí | Sí | Sí | Sí | Sí |
| Dirigido | ||||||
| MLPA | No | No | No | Sí | Sí | No |
| RT-qPCR | No | No | No | Sí | Sí | No |
| FISH | Sí | Sí | Sí | Sí | Sí | No |
Las técnicas de secuenciación de cuarta generación, debido a su capacidad de secuenciar cadenas largas, son las idóneas para la detección y caracterización de las variantes estructurales (Lam et al., 2009; Beyter et al., 2019; Cretu Stancu et al., 2017).
Nuestro grupo ha validado en diferentes enfermedades y con distintos equipos y aproximaciones, la fortaleza de la secuenciación con nanoporos para diseccionar a nivel nucleotídico la totalidad de variantes estructurales.
El estudio de la mayor cohorte mundial de variantes estructurales que causan deficiencia de antitrombina (N=39), una enfermedad rara, monogénica con herencia dominante es un ejemplo perfecto. En este trabajo, diferentes variantes estructurales que afectando el gen SERPINC1 causaban deficiencia de antitrombina, que fueron detectadas empleando diferentes métodos moleculares (MLPA, LR-PCR y secuenciación por la plataforma Illumina o array de CGH), fueron completamente caracterizadas a nivel nucleotídico, definiendo con precisión su extensión y la secuencia de los puntos de ruptura, mediante secuenciación con nanoporos, empleando tanto la plataforma minION como promethION, con o sin enriquecimiento (Figura 4). El estudio muestra cómo la tecnología de secuenciación con nanoporos detecta todas las variantes estructurales independientemente de su tamaño (desde 193 pb a 8 Mb) o tipo (deleciones, o duplicaciones), y ha permitido conocer los puntos de corte de las variantes a nivel nucleótido, permitiendo así demostrar la presencia de elementos repetitivos, así como microhomologías.
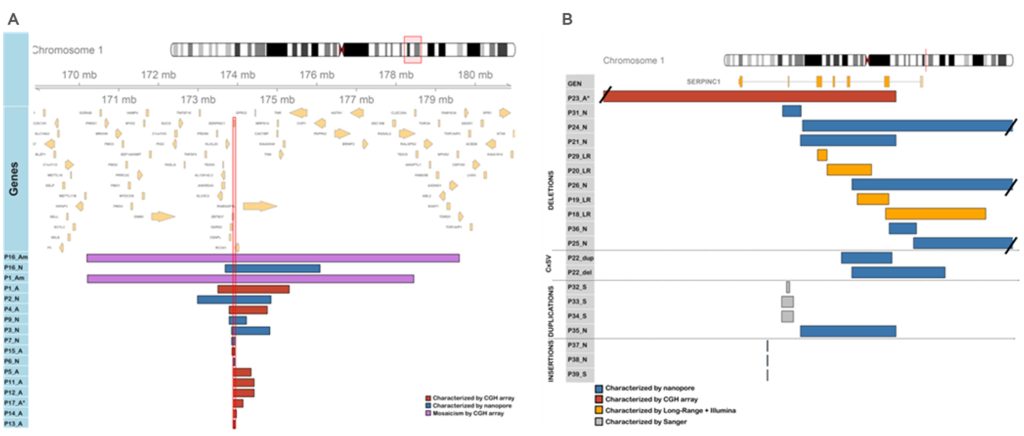
La presencia de estos elementos sugiere la existencia de un mecanismo común basado en replicación (break-induced replication, BIR; microhomology-mediated break-induced replication, MMBIR; fork stalling and template switching, FoSTeS) que involucra elementos repetitivos, para la formación de variantes estructurales causantes de este desorden. Además ha permitido resolver casos con resultados discrepantes obtenidos por otras técnicas (MLPA y LR-PCR), detectando en un caso, una variante estructural compleja, detectada por primera vez en esta enfermedad (de la Morena-Barrio et al., 2022). Otros estudios muestran la fortaleza de esta tecnología de secuenciación por nanoporos para caracterizar variantes estructurales en diferentes enfermedades como autismo (Eisfeldt et al., 2024), parkinson (Daida et al., 2024), deficiencia de FXI (de la Morena-Barrio B et al., 2023) o enfermedades neoplásicas (Bravo-Perez et al., 2024).
La secuenciación por nanoporos también ofrece la capacidad de resolver a nivel nucleotídico unas variantes estructurales complejas como son las expansiones de repeticiones nucleotídicas que se suelen diagnosticar por técnicas basadas en Southern Blot y no siempre se obtienen resultados definitivos. La tecnología de secuenciación por nanoporos está permitiendo definir con precisión las repeticiones (tanto número como secuencia), así como diferenciar los haplotipos permitiendo un diagnóstico más preciso (Wendlandt et al., 2024, Mohren et al., 2024, Van Deynze et al., 2024) .
Estudio de casos de base molecular desconocida
En numerosas patologías existe un porcentaje de casos con fenotipo claro pero con base molecular desconocida en los genes candidatos. Diferentes mecanismos y factores pueden justificar estos resultados, como las alteraciones genéticas en regiones regulatorias o la implicación de otros genes no asociados previamente con dicha enfermedad. Pero la existencia de alteraciones genéticas no detectadas debido a las limitaciones de las técnicas de secuenciación de lecturas cortas es quizás la causa más probable. El uso de la secuenciación de cuarta generación puede cubrir este hueco e identificar variantes de difícil diagnóstico por otras técnicas, como pueden ser: translocaciones atípicas, variantes estructurales complejas o inserciones de grandes elementos repetitivos.
El uso de la secuenciación por nanoporos en distintos casos sin diagnóstico de distintas patologías ha permitido detectar el mecanismo genético causante en algunos de ellos, destacando la detección de inserciones de retrotransposones. En nuestro grupo, hemos reportado inserciones de elementos ALU (Fernández-Suárez et al., 2024) y elementos SVA (de la Morena-Barrio B et al., 2022) (Figura 5). Son ejemplos de enfermedades causadas por la inserción de retrotransposones, diagnosticadas gracias a la secuenciación por nanoporos y que demuestran tanto la dificultad diagnóstica para su identificación como que este puede ser un mecanismo patogénico subestimado y posiblemente implicado en muchas más enfermedades. Además, el estudio de elementos móviles en el genoma humano ha demostrado que el mapa de retrotransposones tiene que definirse con precisión, discriminando variaciones polimórficas de aquellas con potencial impacto patogénico (Cuenca-Guardiola et al., 2023).
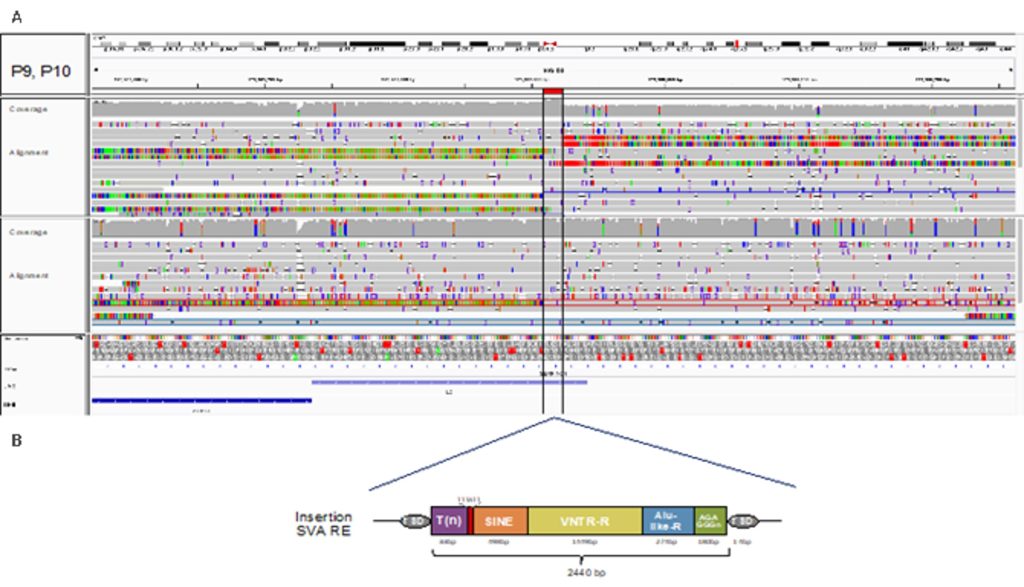
Finalmente, la resolución nucleotídica obtenida por la secuenciación por nanoporos ha permitido definir de qué tipo de retrotransposon se trata y poder realizar estudios filogenéticos, aunque la mayor utilidad de conocer la secuencia nucleotídica de la inserción del retrotransposon es la de permitir el diseño de primers específicos para poder hacer estudios de validación, familiares o poblacionales mediante PCR (de la Morena-Barrio B et al., 2022).
Secuenciación dirigida
La secuenciación mediante nanoporos no solo se puede realizar de ADN o ARN sin manipular, sino que también se puede dirigir a regiones específicas de interés y así optimizar la secuenciación, reduciendo el uso de la celda de flujo y generando librerías de distintas muestras, reduciendo los costes de la secuenciación, o incrementando la profundidad de secuenciación en regiones específicas del genoma.
Existen distintas maneras de dirigir la secuenciación: mediante PCR convencional o LR-PCR, que va a permitir generar gran profundidad de lectura en la región amplificada y mediante enriquecimiento informático in silico (Martin et al., 2022).
Esta última aproximación, es la aplicación más novedosa de esta tecnología, y es posible debido a la capacidad de secuenciar en tiempo real. Gracias a esta propiedad, se puede seleccionar una región o regiones genómicas de interés (0,1-1% del genoma), para que el secuenciador seleccione o descarte las lecturas que coincidan con la región indicada. Esta selección (enriquecimiento, o depleción) se realiza informáticamente en cada uno de los poros que secuencia las cadenas de ADN que se introducen, mediante una API que se ejecuta en el software de análisis de nanoporos: MinKnow.
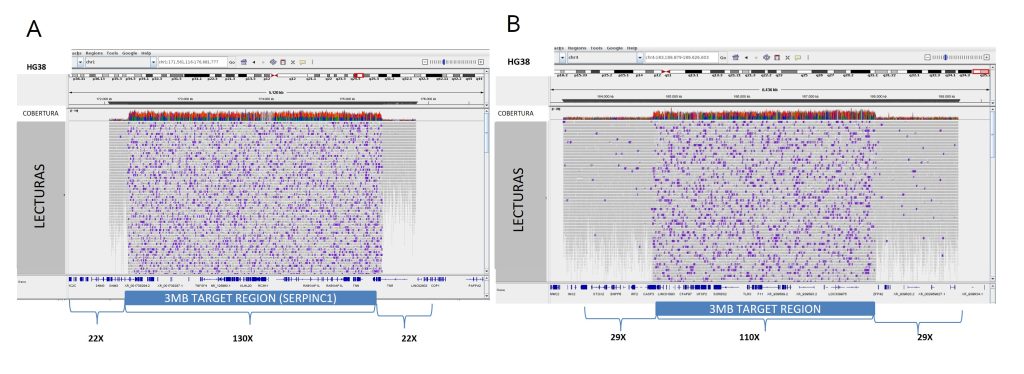
Cualquiera de estas estrategias puede ser de gran utilidad para aprovechar las propiedades de la secuenciación por nanoporos, cuando se tienen genes candidatos. Nuestro grupo tiene experiencia en el uso de enriquecimiento in silico para la identificación de variantes estructurales en genes candidatos. Hemos realizado estudios enriqueciendo 3 Mb incluyendo distintos genes candidatos como: el gen SERPINC1, F11, PAH o incluso panel de genes: 20 genes implicados en cáncer de mama o 12 genes implicados en albinismo (Figura 6).
Definición de haplotipos complejos
Determinar el haplotipo ligado a un alelo, o el ligado a una mutación puede tener diferentes utilidades. La identificación de dos mutaciones tiene diferentes consecuencias patológicas dependiendo de si se localizan o no en el mismo alelo. La determinación de variantes bialélicas, es sencilla con secuenciación de cadenas largas (ya sea de genoma completo o secuenciación de PCR, o de enriquecimiento dirigido) y tiene implicaciones en tratamiento, pronóstico y seguimiento de diferentes enfermedades.
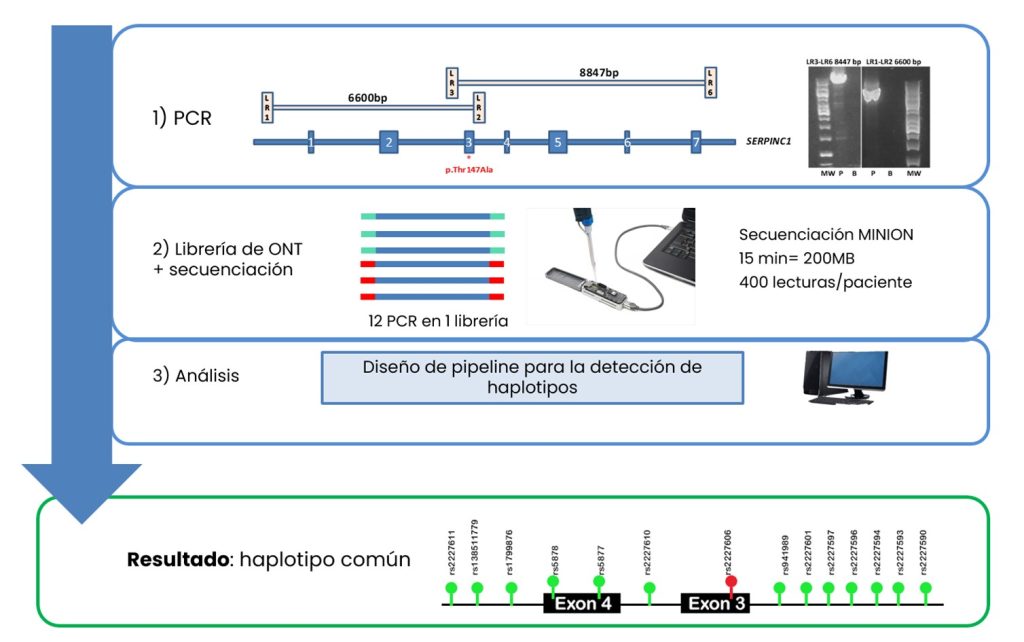
La determinación de haplotipos largos también tiene utilidad evolutiva, para la definición de variantes fundadoras de determinadas poblaciones o para establecer el origen materno o paterno de las variantes de novo entre otras aplicaciones. Este tipo de estudios se escapa a la secuenciación con lecturas cortas y requiere de otro tipo de técnicas como estudios familiares, o clonación, aunque la secuenciación con nanoporos permite una resolución sencilla y rápida, como nuestro grupo demostró para validar el efecto fundador de la variante p.Thr147Ala de SERPINC1, presente en 14 pacientes de distintos países africanos con deficiencia de antitrombina, mediante un método simple, rápido y económico que definió el haplotipado intragénico del alelo portador de esta mutación. La secuenciación de 2 grandes PCR que solapaban la mutación en los pacientes portadores ha permitido definir 13 marcadores ligados a esta mutación y presentes en el alelo mutado de todos los portadores de la variante p.Thr147Ala, demostrando su efecto fundador en población africana (Figura 7) (Orlando et al., 2020).
Epigenética y epitranscriptómica
Al secuenciar material genético sin manipular, la secuenciación por nanoporos identifica cada una de las modificaciones de las bases nitrogenadas, ya que éstas producen distintos cambios en la corriente iónica, que se recoge en el “rawdata”. Hasta la fecha, se puede discriminar las modificaciones epigenéticas tanto de ADN como de ARN, incluyendo: pseudouridina, N6-methyladenosina (m6A), 5-methylcytosina (5mC), y 7-methylguanosina (m7G) (Simpson et al., 2017; Wongsurawat et al., 2022). Se consigue en el mismo proceso datos de secuencia y epigenéticos de cada alelo, generando información de gran valor que ya está teniendo repercusión clínica.
Transcriptómica
La secuenciación de ARN sin manipular ni fragmentar, permite la secuenciación de transcritos completos, mediante lecturas largas. Esta característica hace que la secuenciación de ARN por nanoporos sea una aproximación de secuenciación de ARN y transcriptómica muy potente y el método idóneo para la identificación y cuantificación de transcritos alternativos. Además, este sistema de secuenciación de ARN determina la extensión de la cola de poli-A y detecta variaciones epitranscriptómicas, cuyo impacto funcional se está empezando a estudiar (Mock et al., 2022; Leprêtre et al., 2024; Marquez et al., 2024).
Limitaciones
El punto débil de la secuenciación por nanoporos es la cobertura del genoma (o el precio que hay que pagar por conseguir una alta cobertura). Aunque como hemos comentado existen sistemas de enriquecimiento, sería deseable que estos procesos fueran más económicos y rápidos para conseguir la globalización de esta tecnología.
Nuevas perspectivas
Actualmente se están desarrollando nuevas posibilidades de la secuenciación por nanoporos. Destacamos el estudio de la estructura tridimensional del ADN mediante un protocolo basado en captura conformacional de la cromatina y la secuenciación de lecturas largas. Esta aproximación permite ver el estado tridimensional de la cromatina en el núcleo celular, lo que permitirá estudiar las regiones distales y proximales del genoma, lo que puede identificar elementos reguladores distales de muchos genes y su potencial implicación patológica (Deshpande et al., 2022).
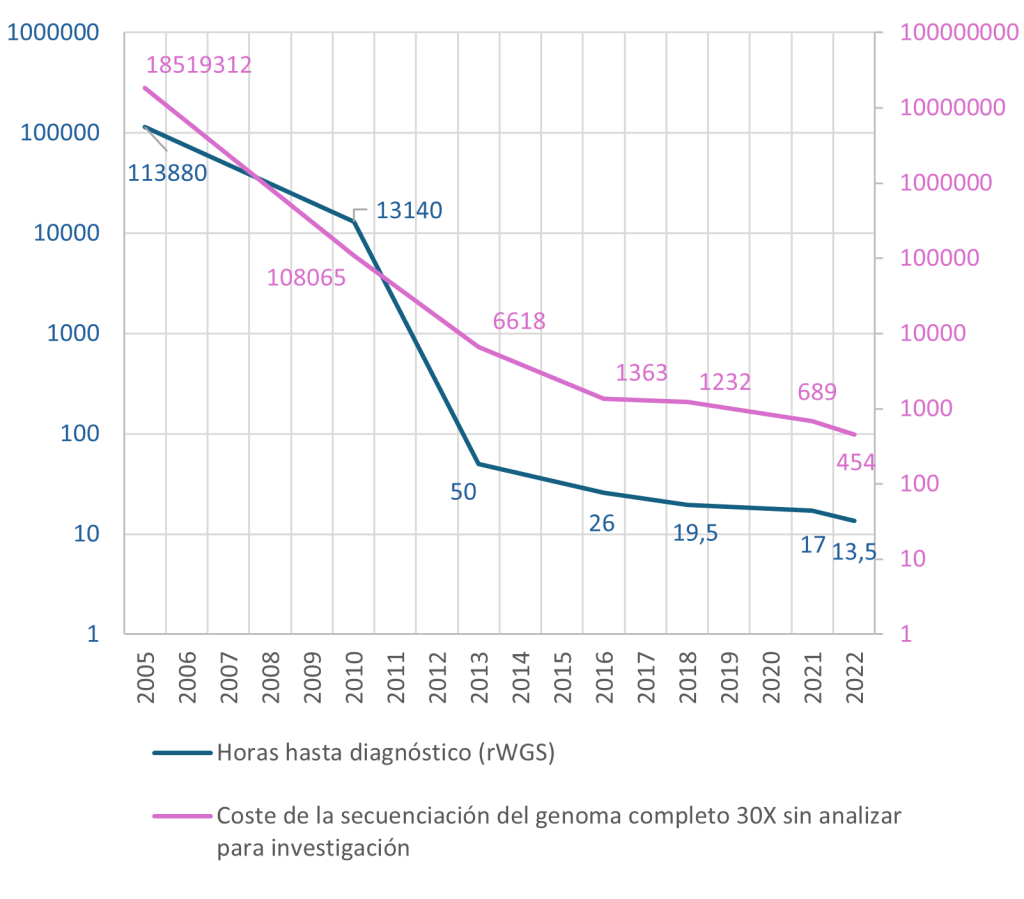
También tenemos que comentar la posibilidad de que la secuenciación con nanoporos pudiera aplicarse al campo de la proteómica. Aunque todavía con resultados muy preliminares se está estudiando el uso de esta tecnología en la secuenciación de proteínas. Finalmente debemos destacar el esfuerzo por conseguir un proceso de secuenciación y sobre todo de análisis bioinformático muy rápido, combinando diferentes mejoras bioinformáticas de alineamiento y análisis de datos. Estos avances han hecho que sea posible obtener un resultado de la secuenciación completa del genoma humano en menos de 10 horas desde que se extrae la muestra de sangre al paciente (Gorzynski et al., 2022).
CONCLUSIONES
La secuenciación de cuarta generación mediante nanoporos ofrece numerosas aplicaciones para el abordaje del diagnóstico molecular, con aplicaciones tanto clínicas como básicas. Destacamos su utilidad en: 1) la completa caracterización de variantes estructurales; 2) la identificación de la base molecular de casos de difícil diagnóstico molecular mediante técnicas convencionales; 3) la resolución de grandes haplotipos de manera sencilla (lo que permite realizar estudios de origen de mutaciones, o el diagnóstico de mutaciones bialélicas); 4) el estudio epigenético tanto de ARN como de ADN; 5) la caracterización de transcritos alternativos.
Finalmente, la secuenciación mediante nanoporo es un campo que puede abrir nuevas perspectivas en la base molecular de enfermedades de base genética, es relativamente sencilla y no requiere de grandes equipamientos para su desarrollo.
Declaración de conflicto de intereses
Belén de la Morena-Barrio, José Padilla Ruiz, Esther Navarro Manzano, Jesualdo Tomas Fernández-Breis y Mª Eugenia de la Morena Barrio ofrecen servicios de secuenciación por nanoporos en la spin off Longseq Applications SL.


