La heterocromía es una anomalía del iris en la que un mismo individuo presenta diferentes colores en sus ojos. Es cierto que esta condición puede resultar muy estética. No obstante, la heterocromía puede aparecer en ocasiones como un síntoma de alguna enfermedad, como en el caso del síndrome de Waardenburg.

El síndrome de Waardenburg es una patología genética con una incidencia en la población de 1/42000 nacimientos, cuyas principales características son:
- Decoloración del flequillo
- Raíz nasal ancha
- Surco subnasal corto
- Heterocromía
- Pérdida de audición
Esos son los síntomas generales, pero cabe destacar que este síndrome puede clasificarse en cuatro tipos: los tipos I, II y III presentan una herencia autosómica dominante, mientras que el tipo IV se transmite de forma autosómica recesiva. Los tipos I y II son los más comunes. En cada tipo los genes afectados son diferentes. Además pueden sufrir distintos tipos de mutaciones y en lugares diferentes de un mismo gen, por lo tanto, varía la sintomatología. Sin embargo, todos ellos son genes que afectan a la función de la cresta neural. También varía la severidad de los síntomas, desde anomalías en la pigmentación hasta deformidades en las extremidades y disfunciones neurológicas. Aunque es una enfermedad crónica, la esperanza de vida es normal.
Desarrollo embrionario: migración de melanocitos desde la cresta neural

Durante el desarrollo embrionario, algunas células de la cresta neural se diferencian a melanocitos. Este proceso depende de los factores de transcripción SOX10, SNAI2, MITF y PAX3, que se encargan de mediar la expresión de varios genes implicados en la diferenciación de los melanocitos. Al diferenciarse, los melanocitos adquieren funciones de pigmentación, es decir, generan pigmentos para el pelo, los ojos y la piel. Así pues, cuando hay mutaciones en algunos de los genes que codifican para los anteriores factores de transcripción, las consecuencias pueden ser enfermedades cuyos síntomas incluyen defectos en la pigmentación. Un ejemplo es el síndrome de Waardenburg, pues algunos de sus síntomas son la heterocromía y la despigmentación en el pelo y la piel.
Las mutaciones que se han encontrado en pacientes con el síndrome de Waardenburg son diversas: inserciones, deleciones, y mutaciones puntuales. En los tipos I y III el gen mutado es el PAX3, y en el tipo II son SNAI2, SOX10 y MITF. Como se ha mencionado anteriormente, los cuatro son factores de transcripción importantes para el desarrollo de los melanocitos desde la cresta neural primitiva, y también están relacionados con la audición. En el tipo IV se han encontrado mutaciones en los genes EDN3, EDNRB y SOX10. Por su parte, EDN3 y EDNRB codifican para moléculas señalizadoras que intervienen también en el desarrollo de los melanocitos.
A continuación, vamos a ver las diferencias en el fenotipo según los genes afectados, para lo que describiremos los cuatro tipos de síndrome de Waardenburg por separado.
Síndrome de Waardenburg tipo I
En pacientes con el síndrome de Waardenburg de tipo I (WS1) las características clínicas que se observan son pérdida de audición, anomalías en la pigmentación de iris, piel y cabello, y distopia cantorum, que es un aumento de la distancia habitual entre los ojos. La pérdida de audición puede ser unilateral o bilateral, aunque suele ser bilateral en la mayoría de los casos, y no es progresiva: los pacientes ya nacen con un determinado grado de sordera y no aumenta con el paso del tiempo.
Diagnóstico
Cuando se observan estas características fenotípicas en un individuo, para confirmar que se trata de WS1 se debe identificar mediante test genéticos moleculares una variante patogénica del gen PAX3, en heterocigosis. Sin embargo, puede ser que los rasgos fenotípicos por sí solos sean concluyentes. De hecho, existe un criterio para este diagnóstico sin test genético, establecido por el Waardenburg Consortium.
En cuanto a la relación genotipo-fenotipo, no se ha encontrado una asociación concreta para la pérdida auditiva. Tampoco se han descrito diferencias significativas en la severidad del síndrome entre individuos que presentan el gen PAX3 totalmente delecionado y los que presentan deleciones parciales. Otra posibilidad es que el paciente sea doble heterocigoto para PAX3 y MITF, en cuyo caso se encuentra un mayor número de anomalías pigmentarias.
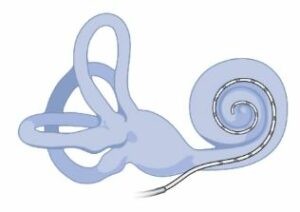
Tratamiento
No hay una cura como tal para la enfermedad, pero sí que se pueden tratar algunos de los síntomas. Por ejemplo, en casos de pérdida de audición severa el implante coclear ha resultado ser muy efectivo. También es recomendable proteger las áreas más afectadas por la despigmentación, utilizando crema solar en las áreas depigmentadas de la piel.
Síndrome de Waardenburg tipo II
Es muy similar al WS1, sin embargo en el WS2 no hay distopia cantorum y la sordera es más frecuente. Además, hay 5 subtipos de esta enfermedad, según el gen afectado y el tipo de mutación. El tratamiento es el mismo que en WS1.
Diagnóstico
De igual manera que en el WS1, puede que las características fenotípicas sean concluyentes. De no ser así, para confirmar el diagnóstico se detectan variantes patogénicas de los genes SNAI2, MITF y SOX10.
Síndrome de Waardenburg tipo III y IV
Estos tipos son muy poco comunes. El WS3 es muy parecido al WS1, con la diferencia de que en WS3 hay deformidades en las extremidades. El WS4 se caracteriza por presentar problemas intestinales.
Gracias a patologías como esta nos damos cuenta de la gran importancia del desarrollo embrionario, pues pequeñas anomalías durante este periodo pueden tener grandes consecuencias en el individuo durante el resto de su vida.
Fuentes:
Ahmed jan N, Mui RK, Masood S. Waardenburg Syndrome. 2020 Sep 25. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan–. PMID: 32809714.
Liu XW, Wang SY, Xing ZK, Zhu YM, Ding WJ, Duan L, Cui X, Xu BC, Li SJ, Guo YF. Targeted next-generation sequencing identified a novel variant of SOX10 in a Chinese family with Waardenburg syndrome type 2. J Int Med Res. 2020 Nov;48(11):300060520967540. doi: 10.1177/0300060520967540. PMID: 33251892; PMCID: PMC7708717.
Milunsky JM. Waardenburg Syndrome Type I. 2001 Jul 30 [updated 2017 May 4]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. PMID: 20301703. https://www.ncbi.nlm.nih.gov/books/NBK1531/
Shelby MV. Waardenburg Syndrome Expression and Penetrance. J Rare Dis Res Treat. 2017;2(6):31-40. Epub 2017 Dec 10. PMID: 30854529; PMCID: PMC6404762.
Si te ha gustado esta entrada y te interesa la genética, descubre nuestros cursos y formación universitaria.