INTRODUCCIÓN
La implementación de las técnicas de hibridación de genoma completo ha optimizado muy notablemente la eficiencia diagnóstica y detección de anomalías cromosómicas, detectando y caracterizando las mismas y superando ampliamente la eficiencia del cariotipo convencional (Dhillon 2014). Actualmente, se acepta la utilidad clínica de esta tecnología de arrays de polimorfismos de nucleótido único y variantes en número de copias (SNPs/CNVs) como base del algoritmo diagnóstico genético para los pacientes afectados de discapacidad intelectual, así como del protocolo para el cribado de pacientes que además han desarrollado fenotipos sindrómicos. Los resultados
obtenidos por este tipo de matrices definen exhaustivamente las alteraciones citogenéticas detectadas por técnicas convencionales, pero sobre todo de un gran número de nuevas pérdidas y ganancias cromosómicas crípticas implicadas en este grupo de patologías. El aumento progresivo de la cobertura de sus diseños, ha ido permitiendo la caracterización de nuevas anomalías cromosómicas subcrípticas, y su posterior correlación fenotípica con otros cuadros clínicos sin etiología conocida. La consecuencia inmediata de la aplicación rutinaria de esta tecnología, ha señalado una serie de nuevos genes de susceptibilidad candidatos a ser valorados como bases moleculares causales del desarrollo de discapacidad mental y/o rasgos dismórficos.
La descripción de los signos y síntomas clínicos observados en los pacientes con fenotipos extremos, se realiza actualmente de forma homogénea en base a un esfuerzo ingente realizado para denominar cada uno de ellos según la base de datos Human Phenotype Ontology (HPO); cada uno de los pacientes afectados exhibe un amplio y complejo conjunto propio de signos y síntomas clínicos, debido al desarrollo individual de distintas anomalías en órganos y rasgos físicos. En relación con esto, a pesar de que los arrays de SNPs/CNVs se aplican homogéneamente como herramienta diagnóstica esencial de búsqueda de la causa de discapacidad intelectual idiopática en niños (Shaffer 2007; D’Angelo 2012), la caracterización fenotípica sigue siendo aún somera y difícil de catalogar. La descripción de variantes causales progresa exponencialmente, constituyendo una valiosísima base de datos que detalla la enorme variedad de desequilibrios cromosómicos nuevos y únicos descritos. El valor de su información se basa en su aplicabilidad al ámbito clínico y su asociación definitiva con alteraciones clínicas no filiadas previamente.
En este artículo presentamos el caso clínico de una niña con un conjunto complejo de signos y síntomas, entre los que destaca un evidente déficit del crecimiento, asociados fundamentalmente a la haploinsuficiencia de los genes OMIM: CTDP1, TNFRSF11A, MC4R, PIGN, CDH19, TMX3, RTTN, ZNF407, TSHZ1, MBP y SALL3.
Para el diagnóstico de la paciente se consideró el uso del array de hibridación genómica como método de primera elección, para la descripción rápida y precisa de las posibles anomalías cromosómicas existentes. Los resultados obtenidos definieron la correlación del genotipo con el fenotipo observado. Sin embargo, el uso de cariotipo convencional fue crucial para entender la complejidad de la alteración que portaba la paciente, así como la etiopatogenia de los grandes reordenamientos citogenéticos y mosaicismos que presentaba. Fue de especial interés la detección y descripción citogenética de parte de la alteración existente en la línea celular portadora del cromosoma 18 en anillo, cuyo porcentaje fue calculado menor al 10%.
DESCRIPCIÓN DEL CASO CLÍNICO
Se presenta el caso de una niña de 6 meses que fue remitida para asesoramiento clínico y posterior tratamiento, siendo la segunda hija de padres no emparentados, jóvenes y sanos. No existían antecedentes familiares destacables. El embarazo fue controlado, terminando con un parto vaginal a las 36+6 semanas de gestación. El peso de la paciente al nacer fue 2.240 g (P10), una talla de 44 cm (P10-25) y un perímetro craneal de 31 cm. La puntuación Apgar fue de 9/9, al minuto y a los cinco minutos respectivamente. En el periodo neonatal, la paciente presentó taquipnea transitoria e ictericia neonatal no inmune que requirió fototerapia de menos de 24 horas. Durante la hospitalización, sufrió una infección por estafilococo y epidermitis séptica, tratada con antibióticos intravenosos sin complicaciones.
Los exámenes físicos de la paciente mostraron hipotonía axial moderada y rasgos dismórficos como hipertelorismo, puente nasal plano, orejas displásicas y pliegues de piel redundantes. A nivel abdominal, presentaba una hernia umbilical leve. La paciente padecía una malformación anorectal con una fístula perineal con abertura anal ectópica con recorrido en horquilla. Mostraba abducciones irreducibles en miembros inferiores y pies planos severos. Se detectó la cavidad septum pellucidum y cavum vergae mediante ecografía. Las emisiones otoacústicas y exploración auditiva del tronco encefálico mostraron potenciales patológicos con déficit de conducción auditiva. La TAC del peñasco del temporal mostró estenosis de ambos conductos auditivos. Se le diagnosticó mediante ecocardiografía un defecto del cierre del septo atrial de tipo ostium secundum y un ductus arterioso persistente.
La paciente llevó audífonos desde los 5 años y vendajes en miembros inferiores, con un programa intenso de fisioterapia. Sufrió un episodio de bronquiolitis leve a los 8 meses y varios episodios de broncoespasmos leves-moderados, con ingreso hospitalario a los 11 y 12 meses de edad. Presentaba un evidente retraso psicomotor (consiguió sostener la cabeza a los 8 meses, mantenerse sentada a los 17 meses y levantarse a los 3 años), retraso en el lenguaje (sólo balbuceaba) e hipotonía. Su estudio cerebral mediante resonancia magnética mostró hipoplasia del cuerpo calloso, hipertrofia de la masa intermedia del tálamo y escasa diferenciación entre la materia gris y blanca. El electroencefalograma fue normal.
Desde los 9 meses la talla baja era manifiesta, midiendo 80 cm a los 2 años (en P1, correspondiendo a -2.35 DE) con un peso por debajo del P1 (9 kg, correspondiente a -2.52 DE). Tenía una microcefalia por debajo del P1 desde el nacimiento (-3.64 DE) con plagiocefalia con asimetría posicional y una asimetría de la cara con menor desarrollo de la parte izquierda. Presentaba además fisuras palpebrales horizontales, ojos pequeños con epicanto antimongoloide, estrabismo en el ojo izquierdo, raíz nasal amplia y deprimida, nariz pequeña en punta y boca pequeña con paladar alto. Presentó micrognatia leve y orejas de implantación baja. El examen del tórax era normal, aunque con incremento de la distancia entre mamilas; el abdomen, la cintura pélvica y las caderas eran normales.
La última evaluación por su pediatra especialista en dismorfología fue a los 4.6 años y mostró mejoría general. Aunque la mayoría de los rasgos dismórficos persistieron, se detectaron cambios en el fenotipo de la paciente como la normocefalia y la mejora de la plagiocefalia asimétrica posicional, ojos normales con epicanto bilateral y mejora de su estrabismo. Entonces se describió la existencia de un surco subnasal corto.
ESTUDIOS CITOGENÉTICOS Y MOLECULARES
Después de obtener el consentimiento informado, se realizó un cultivo de linfocitos de sangre periférica de la paciente y sus progenitores para llevar a cabo el cariotipo de bandas GTG de alta resolución (Figura 1). La alteración cromosómica fue molecularmente caracterizada usando array citogenético de genoma completo 2.7 M de Affymetrix (Affymetrix Inc., Santa Clara, CA, USA) (Figura 1). Los datos fueron recolectados usando Gene Chip Scanner 3000 Dx y los ficheros CEL fueron analizados con el software Chromosome Analysis Suite (ChAS v1.1, Affymetrix inc, Santa Clara, CA, USA). La referencia para la comparativa de datos obtenidos fue hg19. Las CNVs detectadas en la paciente fueron comparadas con la Database of Genomic Variants (DGV; http://projects.tcag.ca/variation) y con el International Standards for Cytogenomic Arrays Consortium (Public ISCA database; https://www.iscaconsortium.org). La Database of Chromosomal Imbalance and Phenotype in Humans (DECIPHER; https://decipher.sanger.ac.uk) fue usada como referencia principal para la evaluación de la posible capacidad deletérea de las alteraciones detectadas.
Se realizó análisis por secuenciación Sanger automática bidireccional de toda la región exónica y secuencias flanqueantes del gen MC4R, a partir de ADN genómico en la paciente. Las secuencias de los cebadores usados para la amplificación fueron diseñadas con el software Primer 3 (Untergasser 2012) (secuencias disponibles bajo solicitud). La secuenciación se realizó en secuenciador ABI 3130 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) usando Big Dye Terminator v3.1 Cycle Sequencing Chemistry (Applied Byosystems, Foster City, CA, USA), de acuerdo con el protocolo recomendado por el fabricante. La asignación de bases fue realizada con el programa Sequencing Analysis v5.2 (Applied Biosystems, Foster City, CA, USA). Las secuencias obtenidas fueron analizadas con los softwares Staden package (Staden 1996) y SeqScape v2.5 (Applied Biosystems, Foster City, CA, USA) y comparadas con la secuencia de referencia (ENSG00000166603).
RESULTADOS
El cariotipo convencional de alta resolución y bandeo GTG evidenciaron que la paciente presentaba dos líneas celulares en mosaico: el 92% de las células portaban un cromosoma 18 anormal con una duplicación de la región 18p y una deleción de la región 18q, y el 8% de las células portaban un cromosoma 18 en anillo. Estas alteraciones cromosómicas fueron molecularmente caracterizadas por el array. La fórmula del cariotipo fue mos 46,XX,der(18)add(18)(q21.3)del(18)(q21.3)dn[46]/46,XX,r(18)(p?q21.32)dn[4], siguiendo la normativa establecida en la International System for human Cytogenetic Nomenclature (ISCN). Por tanto, la duplicación 18p del cromosoma 18 derivado se localiza en 18q21.3 tras sufrir la deleción. El estudio citogenético convencional de ambos padres mostró en ambos un cariotipo normal.
El análisis del array detectó tanto la duplicación de 12.5 Mb en el brazo corto (de chr18:12602631 hasta el telómero) del cromosoma 18 derivado que portaba la línea celular mayoritaria, así como la deleción de 20.3 Mb en el brazo largo (de chr18:57691236 hasta el telómero) observada tanto en la línea mayoritaria (92% células con cromosoma 18 derivado) como en las células portadoras del cromosoma en anillo (calculado en un 8%). La fórmula molecular obtenida con el array de genoma completo fue arr[GRCh37]18p11.32p11.21(chr18:136226_12602631)x3, 18q21.32q23(chr18:57691236_78014123)x1, usando la nomenclatura del ISCN (Figura 1). El resto de los resultados obtenidos fueron normales.
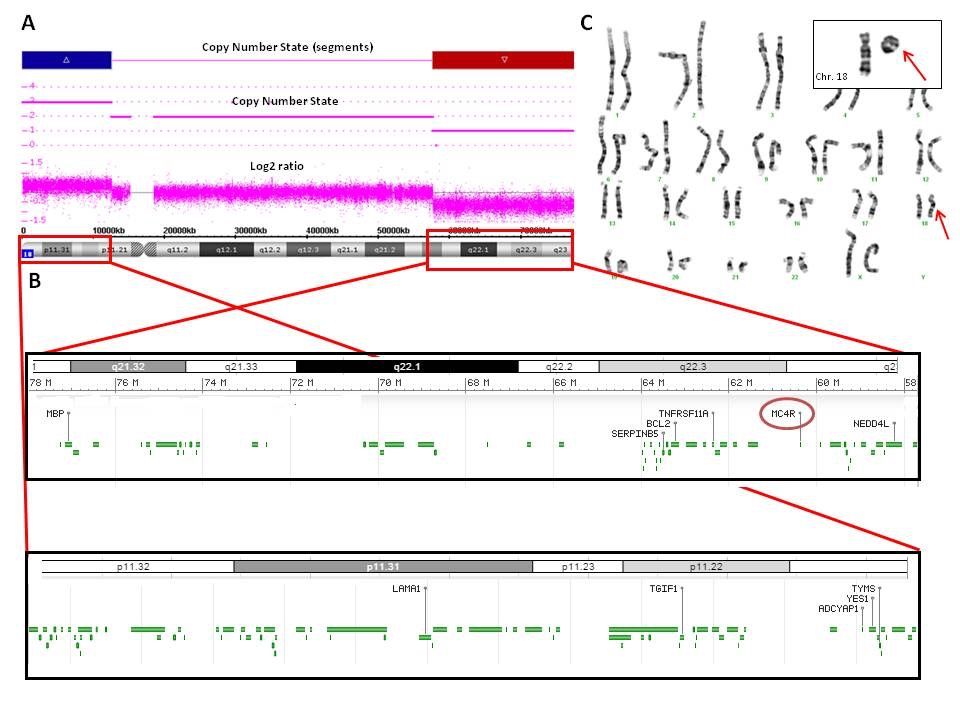
La zona duplicada afectaba 12.5 Mb, que incluía un total de 53 genes. Destacaban los genes con entrada en la base de datos OMIM: NDUFV2, LPIN2, USP14, SMCHD1, TGIF1, APCDD1, GNAL, AFG3L2 y IMPA2.
La deleción mostraba un tamaño de 20.3 Mb y punto de rotura proximal en la posición 57,691,236 del brazo largo del cromosoma 18 (ChAS software (hg19)). En dicha área delecionada se localizaban un total de 32 genes, de los cuales CTDP1, TNFRSF11A, MC4R, PIGN, CDH19, TMX3, RTTN, ZNF407, TSHZ1, MBP y SALL3 tenían entrada en la base de datos OMIM (Figura 1).
Los resultados del array no revelaron datos acerca de la caracterización molecular de otra pequeña deleción distal en 18p, que portaba el cromosoma 18 en anillo. Dicha deleción distal 18p, responsable de originar el cromosoma en anillo, pudo no ser detectada por el array fundamentalmente por su coincidencia con la duplicación de la línea mayoritaria. El porcentaje calculado para esta línea celular (8%), no fue insuficiente para detectar la deleción localizada a nivel del brazo q.
Las secuencias del gen MC4R, cuya alteración está implicada en el desarrollo de obesidad, fueron normales, sin mutaciones conocidas ni otras variantes no descritas. Otros genes como CTDP1, TNFRSF11A, PIGN, CDH19, TMX3, RTTN, ZNF407, TSHZ1, MBP y SALL3 no se consideraron candidatos a ser secuenciados porque su capacidad funcional individual en humanos no está bien caracterizada.
DISCUSIÓN
Presentamos el caso de una niña que mostraba un conjunto de signos y síntomas que resultan en un fenotipo complejo, molecularmente caracterizado por un cromosoma 18 derivado portador de una duplicación de 18p11.32p11.21 (en el 92% de las células) y una deleción en 18q21.32q23, también presente en la línea celular en anillo (en el 8% de las células), que causaba fundamentalmente la haploinsuficiencia conjunta de los genes CTDP1, TNFRSF11A, MC4R, PIGN, CDH19, TMX3, RTTN, ZNF407, TSHZ1, MBP y SALL3. La caracterización de la deleción de novo de 20.3 Mb (arr[GRCh37]18q21.32q23(chr18:57691236_78014123)x1) evaluó además la capacidad deletérea del conjunto de 32 genes contiguos delecionados. El último examen clínico realizado a los 4.6 años confirmó la mayoría de los signos, síntomas y malformaciones aparecidas desde el nacimiento, correlacionándolos con los resultados obtenidos con el microarray.
La deleción 18q fue comparada con otras deleciones similares descritas en otros pacientes, las cuales también incluían el gen MC4R, (Hale 2000; Cody 2009; Margarit 2012; Versacci 2005), permitiendo la correlación genotipo-fenotipo en dicha serie y en nuestra paciente. Los genes delecionados en esta región (de 18q21.32 a 18q23) habían sido previamente asociados fundamentalmente con microcefalia, atresia del canal auditivo y defectos de la mielinización (Hale 2000; Margarit 2012). El retraso psicomotor y de lenguaje, así como el déficit neurológico se ha relacionado también con pérdidas similares de estos genes contiguos (Cody 2007). El gen TSHZ1 está asociado con la atresia auditiva congénita, con ausencia bilateral o formación incompleta del canal auditivo, y también se relaciona con alteraciones en el oído medio (Feenstra 2011). Estos signos se han detallado en aproximadamente el 66% de los pacientes con deleción terminal en 18q (Veltman 2003).
Aunque la obesidad es una patología compleja cuya etiología genética se ha relacionado con un amplio número de genes, se han descrito la haploinsuficiencia (Cody 1999) y el déficit función de MC4R (Abdullah 2016; Farooqi 2005; Turner 2015) como causas monogénicas no sindrómicas mayoritarias. Se conoce muy poco acerca del comportamiento de las alteraciones de los genes de susceptibilidad a obesidad no sindrómica, asociados a desequilibrios cromosómicos; las series analizadas tratan de identificar en los pacientes con discapacidad intelectual asociada a obesidad, unas bases moleculares diferentes (D’Angelo 2012).
No se observaron los efectos esperados por la deleción del gen MC4R en nuestra paciente, quizás por estar contrarrestados por la gran capacidad deletérea de las alteraciones que portaba y que causaban su grave fenotipo sindrómico. Se analizaron en la paciente parámetros específicos para detectar evolución del crecimiento, signos y síntomas de fenotipo metabólico y/o daño neurocomportamental por la haploinsuficiencia del gen MC4R. Sin embargo, la evaluación en la paciente fue normal. Adicionalmente, la secuenciación automática Sanger de este gen confirmó que la paciente no portaba ninguna mutación, mostrando un patrón molecular normal en la única copia del gen que portaba. Por tanto, el no desarrollo de obesidad en la paciente podría deberse al complejo genotipo, compensando la haploinsuficiencia del gen MC4R.
El retraso en el desarrollo fue siempre evidente en la paciente. Entre los pacientes portadores de deleciones terminales 18q desde la banda q21 hacia el telómero, suele describirse una escasa ganancia de peso y el déficit en el crecimiento físico después del nacimiento. Se atribuyen a la hipotonía, al reflujo gastroesofágico y paladar ojival, los cuales pueden ocasionar graves dificultades para succionar, tragar y/o engancharse en el pecho (Hale 2000; Feenstra 2007) (http://www.rarechromo.org/information/Chromosome%2018/18q%20deletions%20from%2018q21%20and%20beyond%20FTNW.pdf). Sin embargo, un porcentaje de los pacientes que llegaron a edad adulta, alcanzaron pesos normales e incluso desarrollaron sobrepeso; algunos autores sí han sido descrito la aparición de obesidad en un número destacable de individuos portadores de 18q- (Feenstra 2007).
Esta anomalía cromosómica fue aún más compleja ya que la paciente además presentaba en el cromosoma 18 derivado (mosaico del 92% de las células), una duplicación de 12.5 Mb del telómero del brazo corto (trisomía) del cromosoma 18, localizada en 18q21.3, y la deleción 18q. La duplicación 18p es una anomalía cromosómica rara. Hay pocos casos descritos en la literatura (Mabboux 2007). La mayoría de pacientes tienen un fenotipo aparentemente normal con pocos rasgos dismórficos y puede manifestarse con discapacidad intelectual de grado variable. La paciente presentó una malformación anorectal consistente en una fístula perineal con ano ectópico y abierto. Se han descrito casos de pacientes portadores de trisomía parcial de la región 18p-q12 y que padecían malformaciones anorectales, sin definirse la región causal implicada (Schramm 2011).
Del conjunto de genes localizados en la duplicación, destaca IMPA2 por su asociación a mayor susceptibilidad a padecer convulsiones febriles (Nakayama 2004).
Parece esencial en este caso la aceptación de que la especie humana tolera mejor el exceso de material genético que su pérdida. Nuestra paciente muestra unos hallazgos clínicos fundamentalmente asociados con la pérdida de material 18q. Sin embargo, la malformación anorectal puede estar más relacionada con la duplicación 18p (Schramm 2011). Como en la mayoría de los pacientes portadores de estas alteraciones complejas, ambos progenitores no presentaron problemas clínicos ni de desarrollo y mostraron tener cariotipo normal; la alteración citogenética compleja fue considerada de novo en la paciente.
Aunque la hibridación de ADN genómico mediante array de alta resolución tiene un papel principal en el diagnóstico de la discapacidad intelectual, el cariotipo convencional fue clave en este caso para identificar el mosaicismo, las reorganizaciones cromosómicas y el cromosoma en anillo. Por tanto, es necesario realizar esta técnica convencional para confirmar genotipos citogenéticamente complejos como el que aquí se expone y postular el mecanismo molecular de origen.
Resumimos cómo la línea celular predominante formada por un cromosoma 18 derivado, que representa el 92% del mosaico, apareció debido a un evento de duplicación de la región 18p localizada en la región 18q21.3, la cual se ha observado y se caracterizado con el cariotipo convencional y el array de genoma completo respectivamente. Este cromosoma derivado además presenta una la deleción en 18q. La línea celular minoritaria, que representa el 8% del mosaico, apareció por dos eventos de deleción, uno en la región 18q distal, el cual es una deleción de gran tamaño caracterizada por el array y también presente en la línea mayoritaria, y una deleción pequeña en la región 18p no detectada por el array debido al porcentaje pequeño de la línea celular así como a la presencia de la duplicación en la línea celular mayoritaria, enmascarando la pequeña deleción responsable del cromosoma en anillo.
CONCLUSIÓN
Utilizando la técnica del microarray de SNPs/CNVs, se ha descrito el fenotipo complejo de una paciente portadora de dos líneas celulares en mosaico, siendo la mayoritaria la que presenta un cromosoma 18 derivado con una duplicación 18p y una deleción 18q y siendo la línea minoritaria la que presentó un cromosoma en anillo originado por una gran deleción 18q y una pequeña deleción en la región 18p distal no detectada por el array. En el caso de fenotipos complejos, el cariotipo convencional es esencial para resolver el diagnóstico molecular. La evaluación de los genes OMIM incluidos en la deleción mostró que la haploinsuficiencia 18q correlaciona con la mayoría de signos y síntomas del fenotipo de la paciente. Además, la haploinsuficiencia del gen MC4R no alteró el metabolismo ni se asoció con el desarrollo de obesidad en la paciente. La descripción del genotipo y de las características fenotípicas de la paciente facilitará la evaluación de otros pacientes portadores de alteraciones similares.
CONFLICTO DE INTERESES
Todos los autores reconocen y revelan no tener conflicto de interés que declarar. Este trabajo fue apoyado por ISCIII, Madrid con apoyo financiero (proyectos 07PI594, 10PI357). David Albuquerque es beneficiario de una beca postdoctoral (SFRH/BPD/109043/2015) de Fundação para a Ciência e a Tecnologia (Portugal).
AGRADECIMIENTOS
Agradecemos el conocimiento de Ines Quintela, la persona al cargo de la plataforma de Affymetrix en CeGen, Nodo de Santiago de Compostela, ubicado en el Hospital Clínicos Universitario, dirigido por el Dr. Ángel Carracedo.

