Imagina que un día de 1902 estás diagnosticando a un niño de tres meses de una enfermedad que se cree asociada a una infección gástrica y cuando te vas a dar cuenta eres miembro de la Royal Society de Medicina de Londres y has sido la primera persona en describir una enfermedad genética hereditaria: la alcaptonuria.
Esto es lo que le ocurrió a Sir Archibald Garrod cuando investigó las relaciones de consanguinidad en la familia del niño afectado de alcaptonuria y dedujo que la enfermedad seguía unos patrones como los que dictan las leyes de Mendel. A día de hoy, este hecho nos puede resultar aún más impresionante teniendo en cuenta lo poco que se conocía sobre desórdenes metabólicos y enzimas en la época
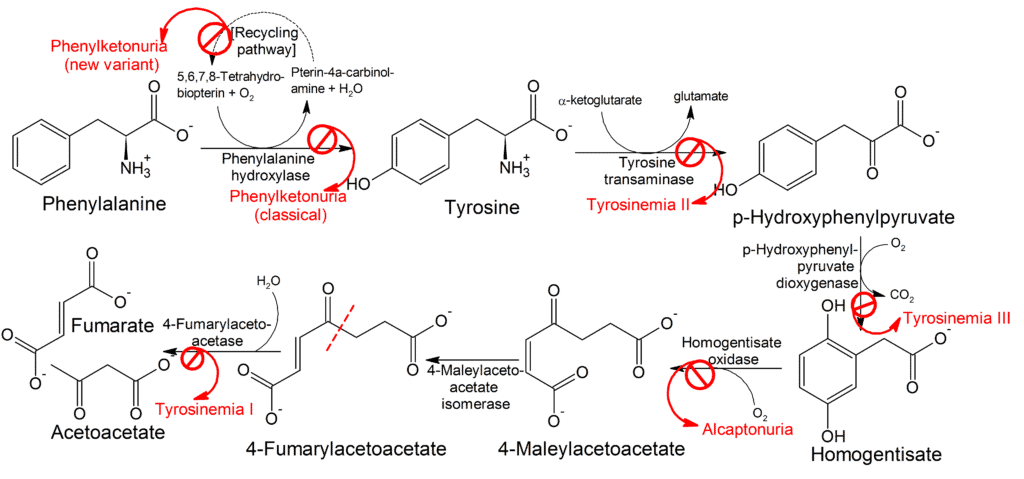
Porque la alcaptonuria (AKU, #203500) es, eso, un trastorno metabólico hereditario. Es una enfermedad autosómica recesiva que puede presentarse en homocigosis (ambas copias del gen responsable están mutadas en el mismo sitio) o en heterocigosis compuesta (ambas copias del gen están mutadas pero en diferente sitio). La alcaptonuria está caracterizada por mutaciones de pérdida de función en el gen HDG, (de las cuales hay más de 120 variantes diferentes descritas en todo el mundo, y la mayoría ocurren en los exones 3, 6, 8, y 13).
Las mutaciones en el gen HDG producen deficiencia de la enzima homogentisato 1,2-dioxigenasa (HGO), que se encuentra principalmente en el hígado y los riñones, lo que lleva finalmente a la acumulación de ácido homogentísico (HGA) y su producto oxidado, ácido benzoquinona acético (BQA).
La enzima homogentisato 1,2-dioxigenasa está implicada en las rutas metabólicas de degradación de la tirosina y la fenilalanina. El ácido homogentísico es un producto de degradación de estas rutas que en la alcaptonuria no puede ser degradado y se acumula, o es excretado en la orina. El ácido homogentísico se oxida y da lugar a benzoquinonas que polimerizan formando un pigmento similar a la melanina, de ahí el color oscuro característico de las secreciones y tejidos. Como añadido, este pigmento puede producir un número incrementado de reacciones redox y la producción de radicales libres, lo que puede causar deterioro de los tejidos.
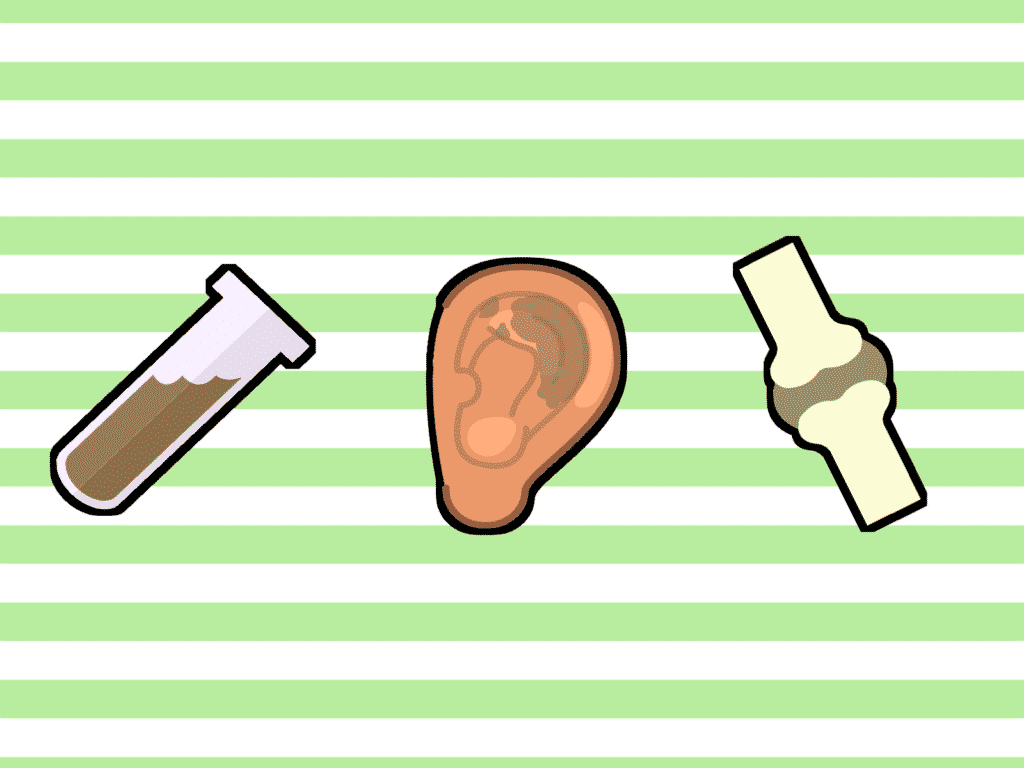
Con excepción de la orina negra en bebés, la alcaptonuria es bastante asintomática en los primeros años y en la juventud. Los otros síntomas que pueden llevar al diagnóstico aparecen ya en la edad adulta cuando la pigmentación oscura empieza a manifestarse en el tejido conectivo de la parte blanca del ojo en forma de manchas oscuras o en el cartílago superior de la oreja (ocronosis).
El principal síntoma de la alcaptonuria es una coloración oscura en ciertos tejidos y secreciones como el cartílago, el sudor, o la más característica, la orina, por tanto esta patología también se conoce como la enfermedad de la orina negra (aciduria homogentísica). A la mayoría de niños menores de un año que padecen alcaptonuria se les diagnostica porque presentan este síntoma.
Por último, el síntoma de la alcaptonuria que se presenta en la edad más avanzada es la artropatía ocronótica, que causa la osificación de las articulaciones de las extremidades, la cadera y la espalda, llegando a ser incapacitante. También caracterizan a la enfermedad el ensanchamiento del tendón de Aquiles, un decrecimiento de la altura significante, una mayor tendencia a fisuras musculares tras esfuerzos leves, una mayor fragilidad de las vértebras y de los huesos largos, afecciones genitourinarias (formación de piedras en próstata y riñón) y afecciones cardiovasculares (calcificación de la arteria coronaria, valvulitis mitral).
La alcaptonuria no afecta significativamente a la esperanza de vida del afectado. La mayoría de tratamientos para la alcaptonuria son paliativos, especialmente para el dolor, ya que no se han desarrollado todavía fármacos verdaderamente efectivos sin efectos secundarios graves para tratar la enfermedad..
Muchos de los pacientes se someten a cirugías para reemplazar articulaciones o válvulas coronarias antes de los 55 años. Además, el dolor se cronifica y el deterioro que causa conlleva la pérdida de movilidad progresiva
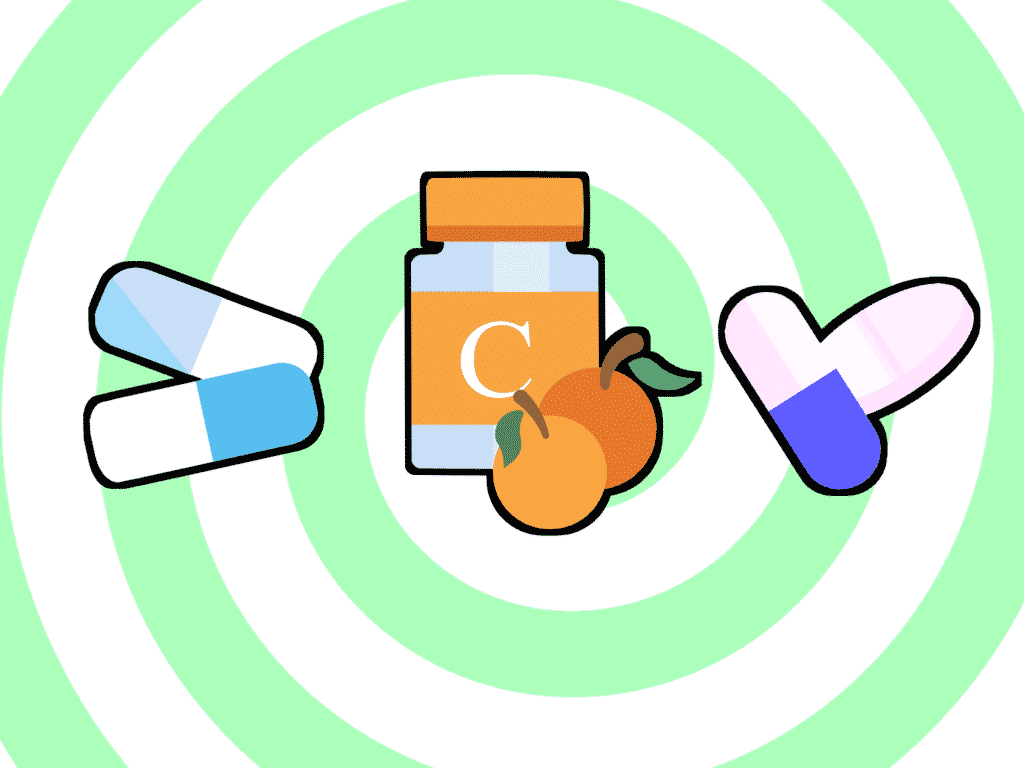
Un tratamiento que se utiliza es la administración de altas dosis de vitamina C como antioxidante para evitar la transformación del ácido homogentísico en productos pigmentados que se acumulan. Sin embargo, en varios estudios se ve que no es muy efectivo, ya que los niveles en sangre se ven muy poco reducidos.
Por otro lado, la terapia más reciente para el tratamiento de la enfermedad que se ha propuesto ha sido el uso del 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione, o nitisinone (Orfadin), que interrumpe la ruta de degradación impidiendo la acumulación de ácido homogentísico,. El problema de los tratamientos con nitisinone es que en lugar de la acumulación de ácido homogentísico se produce el incremento de tirosina en sangre de forma tóxica, con un pronóstico peor que el de la alcaptonuria. De modo que este fármaco puede administrarse siempre y cuando en el tratamiento vaya incluida una dieta estricta sin fenilalanina ni tirosina, o baja en proteínas. Sin embargo, el grado de cumplimiento de ésta es reducido.

Aunque actualmente no haya tratamiento seguro para la alcaptonuria, muchas familias, asociaciones e investigadores están uniendo esfuerzos para lograrlo. Seguro que Garrod no esperaba que su “Errores innatos del metabolismo” y la afirmación de que había enfermedades que se heredaban fuesen a repercutir tanto en el estudio de dichas enfermedades y en los pinitos de la genética en medicina.
¡Nos vemos pronto en otro post!
Referencias
Artivle by David Adam. A father’s fight to help his sons — and fix clinical trials. (2019). Nature International Journal of Science. Volume 565, pages 148-151 . doi: 10.1038/d41586-019-00035-x https://www.nature.com/articles/d41586-019-00035-x
Chanika Phornphutkul. M.D., et al. , Natural History of Alkaptonuria (2002). The New England Journal of Medicine. Volume 347, pages 2111-2121. doi: 10.1056/NEJMoa021736
Mistry J.B., et al. Alkaptonuria (2013). Rare Diseases. doi: 10.4161/rdis.27475 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3978898/
OMIM Alkaptonuria
Original article by Archibald Garrod. Garrod, Archibald E. The Incidence of Alkaptonuria: A Study in Chemical Individuality. (2019) Lancet, Volume II, pages 1616-1620. https://genotipia.com/wp-content/uploads/2019/01/ag-02-1.pdf
Piro Ana, et al. Archibald Edward Garrod and alcaptonuria: “Inborn errors of metabolism” revisited (2010). Genetics in Medicine. Volume 12, pages 475–476 . doi: https://doi.org/10.1097/GIM.0b013e3181e68843
Rovenský J., et al. History. In: Rovenský J., Urbánek T., Oľga B., Gallagher J. (eds) Alkaptonuria and Ochronosis. (2015). Springer, Cham. doi: https://doi.org/10.1007/978-3-319-15108-3_3 https://link.springer.com/chapter/10.1007/978-3-319-15108-3_3



