El diagnóstico genético es determinante para el manejo del Síndrome de DiGeorge, una enfermedad rara causada por una deleción cromosómica y con un cuadro clínico muy variado.
Cada 22 de noviembre, las fachadas de algunos edificios y monumentos se iluminan de color rojo. ¿Sabes a qué es debido? Es un gesto solidario para visibilizar y concienciar sobre el Síndrome de DiGeorge o Síndrome 22q11 en su Día Internacional. ¡Sigue leyendo para descubrir más sobre este síndrome!
¿Qué es el Síndrome de DiGeorge?
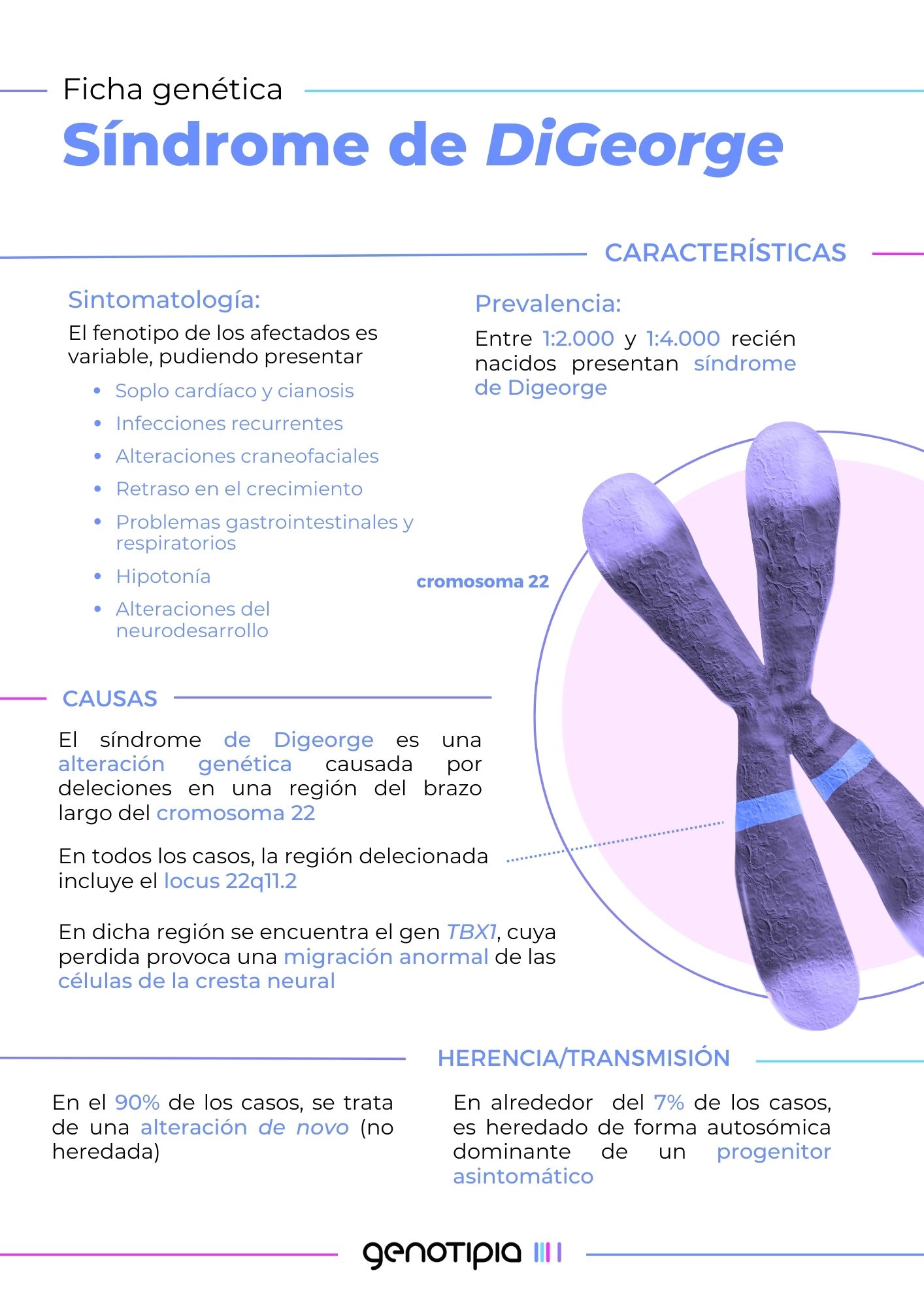
El Síndrome de deleción 22q11.2 o Síndrome de DiGeorge es una enfermedad rara de origen genético que afecta aproximadamente a entre 1/4500 y 1/5000 personas a nivel mundial.
En la década de 1960, el Dr. Angelo DiGeorge, pediatra endocrinólogo estadounidense, realizó un ensayo clínico en niños que presentaban hipoplasia o ausencia de timo y glándulas tiroides, inmunodeficiencia y enfermedades congénitas del corazón. Al analizar los síntomas comunes, el Dr. DiGeorge hipotetizó que todos estos signos podrían estar relacionados, describiendo por primera vez un patrón clínico de síntomas que más tarde se reconocería como el Síndrome de DiGeorge.
Posteriormente, se descubrió que los afectados presentaban una característica genética común: les faltaba un fragmento del cromosoma 22, de entre 0.7 y 3 millones de pares de bases. Dentro de este fragmento genético se encontraban genes relacionados con las malformaciones de los pacientes estudiados. Como reconocimiento de su trabajo en esta patología, tanto la región genética afectada como el síndrome llevan el nombre de DiGeorge.
Manifestaciones clínicas del Síndrome de DiGeorge
El fragmento de DiGeorge que se ve alterado contiene genes que juegan un papel clave en la formación del timo, la glándula paratiroides, el corazón y las estructuras faciales durante el desarrollo embrionario. El número de genes afectados (que no siempre es el mismo en todos los pacientes) determina la gravedad del síndrome de DiGeorge, y por tanto, los síntomas que manifiestan los pacientes, que en general suelen ser:
- Defectos cardíacos como malformaciones en el corazón y grandes vasos, que afectan a la circulación normal de la sangre.
- Anomalías del paladar que pueden dar lugar a dificultades en la alimentación y un habla hipernasal.
- Rasgos faciales sutiles, pero distintivos como párpados algo caídos, ojos más separados de lo habitual o una nariz prominente.
- Inmunodeficiencia y riesgo a sufrir enfermedades autoinmunes como consecuencia de un desarrollo incompleto del timo (más pequeño de lo normal o ausente).
- Niveles bajos de calcio en sangre (hipocalcemia) debido a defectos en el desarrollo de las glándulas paratiroides.
- Problemas en el desarrollo cognitivo: dificultades de aprendizaje y retraso psicomotor.
- Otros: malformaciones gastrointestinales, anomalías dentales y esqueléticas.
La genética del Síndrome: deleción de una parte del cromosoma 22
El fragmento que sufre la deleción se encuentra en la posición 11.2 del brazo largo del cromosoma 22 (22q11.2). Esta región del cromosoma se encuentra delimitada por secuencias repetitivas de ADN muy similares entre sí. Este parecido entre secuencias puede causar confusiones durante el proceso en que el material genético se copia y se divide para formar los óvulos y espermatozoides. Como resultado, a veces se producen errores que provocan la pérdida de un pequeño fragmento del cromosoma 22 (deleción 22q11.2).
La ausencia de esta sección, que engloba hasta tres millones de pares bases, suele producirse de forma espontánea durante la formación de las células sexuales, por lo que se considera una anomalía de novo. Así pues, en el 90% de los casos, la modificación genética no es heredada, sino que aparece por primera vez en el individuo afectado. Solo un 7% de las personas afectadas heredan la alteración genética de alguno de los progenitores, con un patrón autosómico dominante. El resto de casos se deben a mosaicismo en la línea germinal.
Diagnóstico y herramientas genéticas para detectar la deleción 22q11
Cuando se sospecha que una persona podría tener el Síndrome de deleción 22q11, se realiza en primer lugar una evaluación clínica en la que el médico trata de identificar malformaciones características. No obstante, son las pruebas genéticas las que detectan y confirman la ausencia del fragmento del cromosoma 22.
En concreto, entre las herramientas genéticas más empleadas para confirmar el síndrome de deleción 22q11 se encuentra el microarray cromosómico (CMA), capaz de detectar microdeleciones o microduplicaciones genómicas y establecer un diagnóstico diferencial. Otras metodologías complementarias, como FISH, MLPA y qPCR se emplean para confirmar el diagnóstico en casos de sospecha fundamentada.
Para quienes estén interesados en profundizar en estas tecnologías, en Genotipia ofrecemos el Curso Universitario en Técnicas para el Análisis del Genoma, una formación orientada a comprender y aplicar las principales herramientas de análisis genético, utilizadas en el diagnóstico de enfermedades como el síndrome de deleción 22q11.
¿Existe una cura?
Actualmente no existe una cura. Sin embargo, hay abiertas diferentes líneas de investigación para mejorar la calidad de vida de los afectados. Por tanto, el manejo clínico de la enfermedad debe ser multidisciplinar y centrarse en tratar los síntomas específicos de cada persona que puede ir desde la cirugía cardíaca o palatina, hasta la suplementación con calcio o la terapia conductual y de desarrollo.
El tratamiento debe ser lo más individualizado posible dado que la gravedad del síndrome varía en cada paciente y los síntomas no se manifiestan por igual. El uso de herramientas genéticas es de gran importancia para garantizar un diagnóstico diferencial y temprano, ya que en muchos casos identificar el síndrome durante el primer año de vida puede minimizar la intensidad de los síntomas y marcar una diferencia en la calidad de vida.
Para aprender más sobre el ámbito de la Genética Clínica y poder aplicar estos conocimientos a Enfermedades Raras, como el Síndrome de deleción de DiGeorge, la formación especializada de Experto Universitario en Genética Clínica y Enfermedades Raras puede ser muy interesante.
Bibliografía
Orphanet. https://www.orpha.net/es/disease/detail/567
StatPearls. https://www.ncbi.nlm.nih.gov/sites/books/NBK549798/
Asociación España Síndrome 22q11. https://www.22q.es/contenido/35-que-es-el-sindrome-de-delecion-22-q-11.html


