INTRODUCCIÓN
El gen ATXN2 pasaría desapercibido a nuestra atención sino fuera por el segmento repetitivo del trinucleótido Citosina Adenina Guanina (CAG) que se encuentra, frecuentemente, interrumpido por tripletes de CAA. Esta repetición mixta de CAG/CAA se encuentra en el primer exón del gen y codifica un motivo repetitivo de glutaminas (poliQ). El segmento de CAG es inestable y puede llegar a sobrepasar ciertos umbrales en su longitud y con esto causar ataxia espinocerebelosa tipo 2 (aquí SCA2) e incrementar el riesgo para otras enfermedades.
Usualmente existe un determinismo genético donde una mutación causa solo una enfermedad con alguna variabilidad o desviación del fenotipo más común. Sin embargo, la misma mutación en ATXN2 se asocia con varias enfermedades no tan raras, y a la vez causa un amplio espectro clínico en la SCA2. Variaciones puntuales en la secuencia de ATXN2 dan más de “cincuenta tonalidades” a aspectos heredables que van desde el metabolismo de grasas y azucares o nuestra capacidad de discriminar olores, hasta ciertas funciones cerebrales, de ahí que sea un terreno fértil para investigaciones actuales.
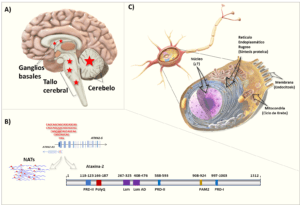
En la SCA2 ocurre fundamentalmente una atrofia progresiva del cerebelo y el tallo cerebral, como resultado de la degeneración y pérdida de las neuronas de Purkinje (Fig.1A). Esto causa un síndrome cerebeloso caracterizado por la alteración de la coordinación de los movimientos, ataxia, dismetría o incapacidad para movimientos exactos, incoordinación para la ejecución de los movimientos de pronosupinación de las manos, temblor, marcha inestable, alteración del habla y enlentecimiento de los movimientos sacádicos oculares. El debut de los síntomas es generalmente a los 30 años como promedio, aunque existe anticipación genética. Esta anticipación implica que los descendientes portadores podrían presentar síntomas de la enfermedad a edad más temprana que la del progenitor afectado. Existe una etapa silente o prodrómica que antecede en años al comienzo de la ataxia, donde a pesar del proceso neurodegenerativo no se detectan cambios evidentes del fenotipo cerebeloso. Sin embargo, hay afectaciones sutiles de la prosodia, la cognición, disautonomía, y problemas con el balance en la marcha y el equilibrio. (Velázquez-Perez et al., 2010).
Existen seis interrogantes centrales para este gen ATXN2 de importancia para varias enfermedades y caracteres humanos (Laffita-Mesa., 2014):
- ¿Cómo se origina la inestabilidad del triplete de CAG de ATXN2?
- ¿Cómo se regula la expresión del gen ATXN2?
- ¿Cuál es su función fisiológica?
- ¿Cuál es el mecanismo patogénico de la proteína ataxina-2 y sus consecuencias en el fenotipo?
- ¿El porqué de la variabilidad fenotípica y el pleiotropismo de la mutación?
- ¿Cómo cambiar del estado mórbido al saludable?
En este trabajo revisamos conceptos generales relacionados con cada una de las preguntas anteriores. También proveemos herramientas actualizadas para poder entender la complejidad genética de ATXN2 de importancia para varias enfermedades humanas. Existe una desproporcionada información acerca de las funciones de ataxina-2 y sus homólogos, pero en este trabajo no pretendemos detallar acerca de esto, si no que más bien trataremos varios temas que no son abordados frecuentemente en la literatura.
OTRAS ENFERMEDADES CAUSADAS POR EL MISMO MECANISMO GENÉTICO DE EXPANSIONES NUCLEOTÍDICAS DE CAG
El mecanismo genético de expansiones de CAG no es exclusivo de SCA2, existen otras 8 enfermedades neurodegenerativas e igualmente incurables causadas por poliQ a saber: enfermedad de Huntington (EH), ataxias espinocerebelosas tipo 1, 3, 6, 7, 17, atrofia dentado-pálido-rubro-lousiana y atrofia muscular espino-bulbar. Como rasgo común en las ataxias espinocerebelosas la neurodegeneración ocurre en el cerebelo y estructuras relacionadas como las vías ascendentes y descendentes de la médula espinal, tanto motoras como sensoriales, afectando la esfera motriz, coordinación e incluso causando síntomas no motrices. En el caso de la EH la degeneración se concentra inicialmente en el núcleo estriado repercutiendo a nivel motor y del comportamiento (DiFiglia et al., 1997). En la atrofia dentado-pálido-rubro-lousiana se degenera una tríada de estructuras centrales que incluye núcleos cerebelosos profundos (núcleo dentado), mesencéfalo (núcleo rubro o rojo) y ganglios de la base (globo pálido). Esto causa un curso desfavorable de la enfermedad con epilepsia, ataxia, movimientos involuntarios, síntomas psiquiátricos y demencia (Koide et al., 1994). En la atrofia muscular espino-bulbar, también conocida como enfermedad de Kennedy, la degeneración de las motoneuronas del tallo cerebral y la espina dorsal resultan en atrofia y debilitamiento muscular progresivo de curso lento, así como contracturas (La Spada et al., 1991).
Todas las enfermedades poliQ, a excepción de la enfermedad de Kennedy, se heredan siguiendo un patrón autosómico dominante, o lo que es lo mismo, los genes defectuosos residen en cromosomas no sexuales o autosomas y con solo una copia del alelo mutante se desarrollará la enfermedad. Para las enfermedades poliQ hay una correlación inversa entre el tamaño de la expansión de CAG y la edad de inicio. Por otro lado, la enfermedad de Kennedy es recesiva y ligada al cromosoma sexual X, por lo que solo los hombres enfermarían al ser XmutY, en tanto que las mujeres XmutX serían portadoras asintomáticas de la mutación. La gran mayoría de las enfermedades poliQ son raras y huérfanas de tratamiento.
GEN ATXN2 Y FUNCIÓN DE LA PROTEÍNA CODIFICADA
El gen ATXN2 fue descubierto al unísono en 1996 por tres grupos independientes de EE. UU., Francia y Japón, los cuales a su vez identificaron la mutación causal de SCA2 usando metodologías distintas: mapeo genético (EE. UU.) uso de anticuerpos monoclonales (Francia) y uso del método de PCR DIRECT (Japón) (Pulst et al., 1996; Imbert et al., 1996; Sanpei et al., 1996).
El gen ATXN2 está en el brazo largo del cromosoma 12 (12q24), se extiende por unas 130kb en el genoma humano y contiene 25 exones con una región promotora bipartita que incluye el primer exón. Este primer exón contiene el segmento repetitivo de CAG. El mismo gen tiene homólogos en otras especies como el ratón, pero en este el motivo poliQ es de solo un residuo (Q) (Pulst et al., 1996; Nechiporuk et al., 1998).
ATXN2 codifica una proteína con función, hasta el momento, desconocida y con amplia expresión en todo el cuerpo humano. En el sistema nervioso tiene una mayor presencia en el cerebelo y la médula espinal. A nivel celular se ha detectado en núcleo, retículo endoplasmático rugoso, membrana celular y mitocondria (Huynh et al., 1999; Auburger et al., 2017). Se ha visto involucrada en el metabolismo del ARN, endocitosis, tráfico del receptor del factor de crecimiento tirosina quinasa, en el metabolismo lipídico y de aminoácidos ramificados, en el de la esfingomielina y en el ciclo de Krebs (Auburger et al., 2017; Sen et al., 2019) (Fig.1BC). Las predicciones iniciales acerca de su función han sido obtenidas del análisis bioinformático de su estructura primaria mostrando varios motivos como PAM, PAM-AD y PR (Albrecht et al., 2004) , los cuales la vinculan como una proteína de unión a ARN (Fig. 1B). Esto está apoyado por hallazgos en modelos de ratón a los que se les ha delecionado (ATXN2-KO) o insertado (ATXN2-KIN) el motivo poliQ anormalmente expandido o el gen humano en un cromosoma artificial.
Se conoce además que ataxina-2 es responsable del cambio de fase de los gránulos de estrés, que son cuerpos de ARN y proteínas, claves para el metabolismo y regulación del ARN que ejercen una función global en la regulación postraduccional cuando hay estrés celular (Auburger et al., 2017; Bakthavachalu et al., 2018). Sin embargo, la función del segmento poliQ aún permanece desconocida, así como los mecanismos patogénicos derivados después de sobrepasar el umbral patogénico. Estudios de asociación recientes señalan que la variación del segmento de CAG -o su análogo en la proteína -poliQ- estaría asociada con la función cognitiva, así como con el volumen de varias estructuras cerebrales i.e. tallo cerebral, putamen, globo pálido, tálamo, y amígdala (Gardiner et al., 2019). Esto a su vez podría explicar la observación de un déficit en el aprendizaje visuoespacial en modelos ATXN2-KO así como la existencia de un tercio de individuos atáxicos SCA2 con déficit cognitivo, y perdida en la discriminación olfativa (Huynh et al., 2009; Valis et al., 2011; Velázquez-Pérez et al., 2006). Por tanto, la perdida crónica de su función o su toxicidad pudiera ser clave a lo largo de la vida para el correcto desarrollo cerebral incidiendo en la cognición y el aprendizaje.
[row]
[col span__sm=»12″ padding=»0px 20px 0px 20px» margin=»0px 0px 0px 0px» bg_color=»#c3c3c3″ bg_radius=»3″ depth=»1″ depth_hover=»1″]
CONCEPTOS CLAVES PARA ENTENDER LA COMPLEJIDAD GENÉTICA DE ATXN2
Gen modificador
Variante genética que interactúa con una alteración genética causal a través de diversos mecanismos en el ARN y proteico. Estos mecanismos pueden cambiar la expresión génica o alterar la función de proteínas influenciando la expresión fenotípica (Génin et al., 2008). En general los modificadores genéticos pueden alterar el fenotipo clínico a través de 4 mecanismos básicos: cambios de la penetrancia, expresividad, dominancia y o la pleiotropía (Davidson et al., 2018).
Penetrancia
Es la ratio de todos los individuos portadores de un alelo mutante respecto a aquellos afectados por la enfermedad. Un ejemplo de penetrancia reducida debido a modificadores genéticos es la retinitis pigmentaria (MIM# 600138). Esta enfermedad es causada por una mutación en el gen PRPF31 (del inglés pre‐mRNA processing factor 31) que típicamente causa déficit visual severo. No obstante, algunos pacientes portadores del alelo causal son asintomáticos. Esto usualmente ocurre debido a una mutación en otro gen CNOT3 (CCR4‐NOT transcription complex subunit 3) que incrementa la expresión de la proteína salvaje de PRPF31 rescatando así el fenotipo mórbido (Venturini et al., 2012).
En el caso de SCA2, este fenómeno se observa en el rango de expansiones 32-35CAG, donde los portadores pueden vivir toda su vida sin mostrar síntomas ni siquiera ligeros de la enfermedad. También, es relativo al instrumento usado para definir el fenotipo mórbido.
Dominancia
Las relaciones de dominancia tales como la autosómica dominante, o la recesiva pueden modificarse por la constitución genética, o sea que alteraciones en otro locus pueden cambiar el efecto de los alelos en el locus causal. Por ejemplo, mutaciones homocigotas (dos alelos mutantes) en el gen HFE regulador de la homeostasis del hierro, causan hemocromatosis hereditaria o sobre carga de hierro (MIM#235200). Sin embargo, los heterocigotos (un solo alelo mutante) desarrollan hiperabsorción de hierro cuando existe mutaciones que causan perdida de función en el gen HAMP (del inglés, hepcidin antimicrobial peptide). Este gen codifica una proteína clave en el mantenimiento de la homeostasis del hierro regulando sus reservas en células macrófagos, así como en la absorción intestinal (Merryweather-Clarke et al., 2003).
Un cambio más sofisticado de esta relación de dominancia fue presentado por Tojima et al., 2018 para ATXN2 y SCA2, la cual es autosómica dominante. Los autores presentaron un caso índice con SCA2 muy leve y de inicio a los 80 años, el cual era homocigoto para dos expansiones de 31CAG y en el que el cribado de secuenciación exómica, así como otras pruebas excluyeron otras alteraciones genéticas. En tanto, su hermano, que fue diagnosticado con ELA de inicio a los 68 y falleció 4 años después por fallo respiratorio, se infiere que era heterocigoto para un alelo intermedio de 31CAG, asociado con ELA.
Como se señala anteriormente, el umbral patológico de SCA2 es ≥33CAG, por lo que 31 tripletes tienen penetrancia nula para SCA2 o sea no causan esta enfermedad. Sin embargo, la presencia de un segundo alelo sugiere un patrón autosómico recesivo para una enfermedad considerada usualmente autosómica dominante (Tojima et al., 2018).
Pleiotropismo
Ocurre cuando el mismo gen tiene dos o más efectos diferentes. El síndrome de Bardet-Biedl (MIM# 209900, 600151, 605231, 615981 615996, 617406,617119) y el de Meckel-Gruber (MIM# 249000, 267010,603194, 607361, 611561, 611134, 612284, 613885, 614175,614209, 615397, 616258) resultan de mutaciones en múltiples genes y se manifiestan con disfunción ciliar. Sin embargo, una mutación en el gen MKS1 (del inglés Meckel syndrome type 1 protein) asociado con el síndrome Meckel-Gruber causa convulsiones en pacientes con el síndrome Bardet-Biedl, aunque estas no forman parte del cuadro clínico de ninguno de los dos síndromes (Leitch et al., 2008).
El gen ATXN2 también se ha asociado con enfermedades muy diferentes a la SCA2 y que necesariamente no se incluyen en el espectro de trastornos del movimiento como lo es la ataxia y el parkinsonismo, ni tan siquiera en la enfermedad de la motoneurona. Por solo citar una de ellas, el glaucoma primario de ángulo abierto es una enfermedad ocular en la que progresivamente se pierde la visión hasta alcanzar ceguera, lo cual no es común en la SCA2. No obstante, una variante intrónica del marcador rs7137828 en ATXN2 se asocia significativamente con esta enfermedad. Incluso se ha detectado una elevada expresión de diversas variantes de procesado alternativo de ATXN2 en la retina y el nervio óptico de afectados (Bailey et al., 2016).
[/col]
[/row]
EXPANSIONES DE CAG EN ATXN2: PROTAGÓNICAS EN VARIAS ENFERMEDADES
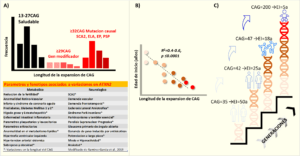
De manera global la longitud del fragmento CAG de ATXN2 no es muy variable, y el rango va desde 13 a 31 CAGs. Como se observa en el diagrama de frecuencias (Fig.2A) 22CAG es el más común (Pulst et al., 1996) y no se asocia con ninguna enfermedad conocida hasta el momento. No obstante, Aguilar et al., 2020 encontraron que la variación dentro del rango normal incrementa el riesgo de obesidad en adolescentes mejicanos (Aguilar et al., 2020).
Se ha propuesto un consenso para buenas prácticas de laboratorio diagnóstico de SCA2 donde las expansiones de 14–31 CAGs se consideran rango normal, las de 32–34 rango de incertidumbre o zona gris, y las de 35–500 expansiones penetrantes (Sequeiros et al., 2010). Sin embargo, es debatible dado el pleiotropismo de esta alteración genética en ATXN2 y a las variaciones de la longitud del CAG en cada región geográfica.
Cuando la longitud del CAG sobrepasa 29 unidades se asocia con esclerosis lateral amiotrófica (ELA), la cual es una enfermedad progresiva, fatal e incurable donde se degeneran las motoneuronas (Elden et al., 2010). Estas controlan los movimientos de los músculos voluntarios encargados de masticar, hablar y caminar. La asociación de ATXN2 con ELA ha sido demostrada en varias poblaciones (Elden et al., 2010; Ross et al., 2011; Laffita-Mesa et al., 2013; Neuenschwander et al., 2014). Incluso el umbral original para ELA se validó, inicialmente, para repeticiones de 24CAG (Elden et al., 2010). Sin embargo, este límite fue refinado para expansiones desde 27-39CAG en varios metaanálisis y estudios de riesgo sugirieron que 30CAG es el valor umbral más específico (Sproviero et al., 2017). El rango de 33-79 tripletes de CAG es causa monogénica de la SCA2, siendo 37CAG el tamaño de expansión más frecuente (Pulst et al., 1996; Imbert et al., 1996; Sanpei et al., 1996). Se han encontrado grandes expansiones de ≥200-884CAG causando síndromes altamente severos con encefalopatía infantil, manifestaciones sistémicas y muerte temprana. (Lastres-Becker et al., 2008). Actualmente, se considera como una extensión del fenotipo de la SCA2.
Existe incertidumbre diagnóstica en el rango de 32-34 repeticiones de CAG (Sequeiros et al., 2010). Esto es porque existen muy pocos casos con un fenotipo neurológico evidente. En el espectro del rango mutante existe superposición genética dado que se han reportado otras enfermedades como la enfermedad de Parkinson (EP), parálisis supranuclear progresiva (PSP), enfermedad de Alzheimer, atrofia multisistema y también demencia frontotemporal (Van Damme et al., 2011). Todo esto sugiere que el segmento repetitivo anormal en ATXN2 vulnera varios grupos neuronales, así como células gliales, a través de la vida y en dependencia del umbral compensatorio de grupos neuronales y de cada individuo, así como de su epigenética se manifestará una u otra enfermedad (Laffita-Mesa., 2014).
En muchos de estos estudios se han identificado otras mutaciones causales, en otros genes, concomitando con las expansiones de CAG en ATXN2 y a la vez otros SNP (del inglés Single Nucleotide Polymorphisms) de menor contribución al desarrollo de neurodegeneraciones (Cady et al., 2014; Dekker et al., 2016), por lo que, si bien la repetición de CAG en ATXN2 no es el factor causal, sí constituye un gen/factor modificador (ver recuadro) incrementando el riesgo a padecer cualquiera de ellas, con preferencia al espectro ELA/demencia frontotemporal y a Parkinsonismo.
Por tanto, debido a esta complejidad genética se propone que la expansión de CAG pudiera influir en el fenotipo de cuatro maneras diferentes: 1) alelos mutantes causantes de SCA2, 2) alelos premutados o de riesgo, 3) alelos dominantes causales de fenotipos no cerebelosos y 4) alelos recesivos causantes de enfermedades cerebelosas en la ancianidad (Pulst, 2018). A estos mecanismos se une uno que recientemente hemos encontrado, consistente en una duplicación en la región digénica ATXN2-S/ATXN2-AS que influye sobre el fenotipo de otras enfermedades en conjunción con los alelos intermedios de ATXN2 (Laffita-Mesa et al., 2020).
Existen otros SNPs que se asocian con glaucoma (Bailey et al., 2016; Rong et al., 2017) y longevidad (Fortney et al., 2015) entre otros (Fig.2A). Por tanto, de todo esto podemos concluir que en relación al gen ATXN2, las expansiones del triplete de CAG son el principal mecanismo genético modificador del fenotipo de varias enfermedades humanas.
LA SINERGIA DEL MENSAJE OPUESTO: ATXN2-ANTISENTIDO
A día de hoy se conoce otro gen que está parcialmente superpuesto con la región 5’UTR/exón/intrón-1 de ATXN2 y cuya orientación está en dirección contraria (o sea 3´– 5’) (Fig.1B) (Li et al., 2016). Este gen, ATXN2-AS, expresa una especie de ARNs conocidos como Natural Antisense Transcripts (NATs) y a diferencia de otros genes el mensaje de ARN no se traduce a proteínas, sino que es un ARN con funciones reguladoras de la expresión génica.
El ATXN2-AS contiene un segmento CUG que es el análogo al CAG, pero en el ARN. Estas moléculas funcionan suprimiendo la expresión del gen que está en dirección 5’ – 3´ (Pelechano y Steinmetz, 2013). De esto se infiere que su expresión podría contribuir a reducir la expresión de ATXN2-S y por tanto disminuir la toxicidad de la poliQ. En el estudio original no se muestran evidencias de esta función y nosotros recientemente no hemos encontrado diferencias en la expresión de estos dos genes que validen una posible regulación (Laffita-Mesa et al., 2020). Esto es además apoyado por la ausencia de coincidencia y nivel de expresión con el patrón de ATXN2 en cerebro y en todo el cuerpo (gtexportal.org).
No obstante, se sabe que en materiales post mortem SCA2, el NAT de ATXN2-AS forma focos en los que se secuestran proteínas esenciales para el metabolismo del ARN, como el regulador de empalme MBNL1. Esto a su vez deriva en trastornos del procesado alternativo del precursor de la beta amiloide (implicado en la enfermedad de Alzheimer) y del gen codificante del receptor de aspartato (Li et al., 2016). Por lo que si bien no se ha demostrado regulación sobre el ATXN2-S, su expresión parece sinergizar la patogénesis del mensaje 5’ – 3´ en SCA2 y otras neurodegeneraciones asociadas con expansiones de CAG en este gen.
INESTABILIDAD GENÉTICA DE LA EXPANSIÓN DE CAG EN ATXN2, MUTACIÓN SCA2
La mutación SCA2 tiene una naturaleza dinámica dado que su longitud puede variar al trasmitirse de padres a hijos. Este es el fundamento de la anticipación genética, en la cual los descendientes portadores debutan más temprano con la SCA2 que los padres por un aumento en la longitud del CAG (Cancel et al., 1997). Otra derivación de esto es la correlación inversa que existe entre la longitud del CAG y la edad de inicio: mientras más larga es la mutación antes será el inicio de la ataxia (Fig. 2B-C). Esta correlación explica gran parte de la severidad del fenotipo en SCA2, variando desde 40-60%.
La inestabilidad somática podría influir en la expresividad (ver recuadro) y variabilidad fenotípica de la SCA2 dado que distintas subpoblaciones neuronales u otros tejidos podrían tener un espectro más o menos amplio del repertorio mutacional. A su vez esto podría diferir en la población y generar patrones de neurodegeneración diferente en el cerebro y en todo el cuerpo repercutiendo en el fenotipo (Matsuura et al., 1999) (Fig. 3A). Así, aquellos casos con SCA2 y fenotipo parkinsoniano podrían mostrar mosaicismo somático diferente en los ganglios de la base que aquellos con un fenotipo puramente cerebeloso.
Se especula que el patrón de interrupciones de CAA en la secuencia de CAG pudiera ser clave porque al estar presente cambiaría el mosaicismo somático. Esto podría actuar de activador causando un cambio en el transcriptoma de ATXN2-S/AS a nivel global, y sería interesante saber cuándo comienzan estos cambios, así como los factores de riesgo subyacentes.
Se ha observado la presencia de estas interrupciones de CAA en familias con SCA2 con fenotipo parkinsoniano respondedor a la L-dopa y también en ELA con expansiones intermedias (Charles et al., 2008, Furtado et al., 2005, Van Damme., 2011). Su ausencia, o lo que es lo mismo, CAG puros, es la regla en casos con SCA2 cerebelosa. Al igual que el triplete CAG, el de CAA codifica para el residuo glutamina (Q). Por tanto, la diferencia solo es a nivel de la secuencia y conformación del ADN y ARN repetitivo y no en la proteína ataxina-2.
MUTAGÉNESIS DEL SEGMENTO CAG EN ATXN2
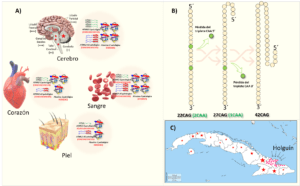
La secuencia del segmento CAG en la población general tiene al menos dos interrupciones de CAA, aunque puede variar. Por ejemplo, para el alelo más común que es el de 22CAG la secuencia es: 8CAG–1CAA–4CAG–1CAA–8CAG. Sin embargo, las expansiones patológicas en SCA2 carecen de estas interrupciones. Son solamente segmentos puros de CAG. Lo anterior es fundamental para entender la mutagénesis en SCA2. Aunque no se conoce al detalle, la mayoría de las observaciones sugieren que la pérdida de las interrupciones de CAA dentro de la secuencia de CAG son un paso clave en la inestabilidad genética de esta secuencia (Choudhry et al., 2001; Laffita-Mesa et al., 2011) (Fig. 3B). En la población cubana se observó que el aumento de la longitud de la secuencia de CAG se asocia con la perdida de la primera interrupción de CAA, ocurriendo una elongación intergeneracional que pudiera generar casos de SCA2 o ELA después de varias generaciones(Laffita-Mesa et al., 2013; Laffita-Mesa et al., 2014).
FACTORES TALES COMO LA EDAD Y EL SEXO DEL PROGENITOR INFLUYEN EN LA TRANSMISIÓN INESTABLE DE LA MUTACIÓN QUE CAUSA SCA2
La edad y el sexo del progenitor influyen en la longitud del segmento CAG de sus descendientes. En el caso de la edad del progenitor se encontró una correlación positiva con el aumento del segmento CAG en la descendencia. Para el caso del sexo la correlación fue que los alelos con 24-31 unidades de tripletes son más inestables cuando la madre es portadora de los mismos. Sorprendentemente, este efecto contrasta cuando los alelos están en el rango causal de la SCA2, donde los alelos paternos son más inestables al trasmitirse a la próxima generación. Lo anterior resulta particularmente contradictorio y puede que el sesgo sea dado porque la baja sensibilidad de la investigación impida detectar el verdadero efecto biológico en el grupo de alelos no patológicos, no así para las expansiones SCA2. Actualmente, no existe evidencia que apoye este cambio de efecto. Sin embargo, para el estudio no se usaron marcadores de haplotipos que permitieran establecer el origen de las trasmisiones entre progenitores-descendientes (Almaguer-Mederos et al., 2018). La variación del fragmento CAG entre progenitores-hijos caía en el rango de error para la técnica de análisis de fragmentos con geles de poliacrilamida (±1.07CAG), y el estudio carece de secuenciación Sanger lo cual sesga enormemente la cuantificación del tamaño del segmento CAG. La gran mayoría de los alelos muestreados provenían de la misma población de casos SCA2 y no se usó una población control independiente para poder contrastar la inestabilidad de alelos largos.
La razón por la que un mismo fenómeno de inestabilidad es influido de manera distinta se desconoce y puede redundar en una falta de datos disponibles para alelos de 24-31 repeticiones CAG, los cuales no son el objetivo de los servicios asistenciales de genética (Laffita-Mesa et al., 2014).
Recientemente, Sánchez-Corona et al. reportaron una familia con el mayor caso de inestabilidad intergeneracional (Sánchez-Corona et al., 2020). En efecto, una mujer de 27 años y con genotipo 22/42CAG trasmitió su hijo una expansión en ATXN2 de ~884CAG. El genotipo del padre es de 22/22CAG y la abuela y tías maternas también están afectadas por SCA2. Esto confirma la segregación de la mutación vía materna que fue trasmitida al hijo.
¿PODRÍA LA FUNCIÓN DE ATAXINA-2 SUBYACER AL EFECTO PARENTAL?
Se especula acerca de por qué los hombres trasmiten alelos más largos a sus descendientes. Las interpretaciones parten de observaciones hechas en el ADN de la sangre por lo que no tenemos una medida exacta de la inestabilidad somática del segmento CAG en las células germinales. Estas últimas son, a la larga, las que portan la expansión de CAG que se trasmitirá a la descendencia. Unido a esto, la sangre es donde menos se expresa ATXN2-S/AS, lo que contrasta con los ovarios y testículos que son de los tejidos donde más se expresa ATXN2-S/AS en todo el cuerpo (gtexportal).
La gran mayoría de las explicaciones se centran en la replicación, reparación, recombinación y mutaciones premeióticas. Sin embargo, ¿cuál es la longitud del fragmento CAG haploide del espermatozoide fecundador dentro de los millones que participan en este fenómeno? Dentro de esta población de gametos, existirá un 50% de espermatozoides con 22CAG y otro 50% con amplio repertorio de longitudes del segmento CAG. Habrá espermatozoides mutantes con más ventajas de supervivencia que otros mutantes e incluso que otros con expansiones normales y esto podría depender, a su vez, de la función y expresión de ATXN2.
En la enfermedad de Huntington, donde también se ha observado este efecto parental, se sabe que el mosaicismo del segmento CAG es mayor en esperma que en sangre (Telenius et al., 1994; Wheeler et al., 2007). Por tanto, a esto subyace la pregunta ¿son aquellos espermatozoides con alelos más largos los más exitosos en la fertilización? ya sea por su mayor frecuencia dentro de la población germinal, y/o porque la pérdida o ganancia de la función de ATXN2 favorece su tránsito hacia fecundar el óvulo.
Las funciones de ataxina-2 estriban en el crecimiento celular lo cual es ventajoso a nivel gamético. Existe correlación positiva entre tamaño de las glándulas sexuales y de espermatozoides con el éxito en la fertilización en mamíferos placentarios (Tourmente et al., 2011). Testículos mayores producen más gametos y más elongados. Estos tienen flagelos más largos, más mitocondrias y cabezas más alargadas, todo lo cual permite nadar distancias mayores hacia el óvulo. En esta línea se sabe que ataxina-2 es esencial para el desarrollo de la línea germinal. Se ha encontrado que la ausencia de ataxina-2 en el gusano C. elegans reduce el número de células germinales y causó masculinización de la línea germinal (Ciosk et al., 2004; Maine et al., 2004). Además, en gusanos sometidos a restricción calórica, los niveles de ATXN2 correlacionan inversamente con el tamaño, contenido graso y desarrollo animal (Bar et al., 2016).
En células humanas uno de los efectos derivados de la perdida/ganancia de la función de ATXN2 incluye la represión de la fosforilación dependiente de mTOR, así como la de PINK1 por lo que con la perdida de las funciones de ataxina-2 se favorecen funciones anabólicas como el crecimiento y la proliferación celular, mayor disponibilidad de lípidos y reservas de glicógeno como alternativas energéticas de alta demanda, así como la síntesis proteica en periodos de estrés celular (Gispert et al., 2009).
La diferenciación del espermatozoide conlleva ensamblaje del flagelo y formación de mitocondrias requiriéndose gran demanda de actina en el lugar y momento oportuno (Teves et al., 2020). Dos estudios indican que la ataxina-2 es necesaria en la alineación correcta de los filamentos de actina en la mitosis, controlando una ruta donde regularía a inhibidores claves de la citocinesis (Gnazzo et al., 2016; Stubenvoll et al., 2016).
También se han encontrado mutaciones en el homólogo de ataxina-2 en mosca (Drosophila Melanogaster) que impedían la unión de esta con la actina y causaron disrupción de los filamentos con la repercusión fenotípica de esterilidad, cerdas aberrantes y letalidad (Satterfield y Pallanck, 2002). Se podría especular que la variación del segmento CAG podría representar una estrategia evolutiva en los gametos que aseguraría éxito en la fecundación mediante la función de ATXN2 y otras proteínas poliQ, no solo por su papel con la actina sino también por su rol en la estabilización y transporte de gránulos de estrés al sitio requerido.
EL CASO INUSUAL DE LA ELEVADA PREVALENCIA DE SCA2 EN CUBA
La SCA2, es una enfermedad rara en el mundo. Sin embargo, alcanza las mayores tasas de prevalencia e incidencia en la provincia de Holguín, Cuba, donde existen más de 500 enfermos vivos y 7173 descendientes en riesgo que pertenecen a ~200 familias (Fig. 3C). La prevalencia supera, desafortunadamente, en más de 20 veces lo encontrado en otros países, y en el mundo. En la provincia de Holguín, este valor es de 45:10000, con una prevalencia mayor en los municipios Holguín, Cacocum y Báguanos (61.90, 78.84 y 141 por cada 10000, respectivamente). Actualmente, los casos están dispersos por la isla lo cual representa un serio problema de salud nacional. Anualmente nacen 22 niños portadores de la mutación, enferman 35 nuevos casos y fallecen 15 enfermos (Velázquez Pérez et al., 2009).
Aunque no existe cura para la SCA2, actualmente se les brinda diagnóstico molecular y tratamiento sintomático con énfasis en la rehabilitación física, lo cual es un alivio considerable para las familias. También se investiga intensamente el fenotipo clínico, así como los mecanismos bioquímicos y moleculares que modulan la enfermedad en las familias afectadas.
PISTAS DE UN EFECTO FUNDADOR EN LA SCA2 CUBANA
Se hipotetiza que la mutación fue introducida por emigrantes hispanos a inicios de la fundación de la villa Holguín. Esto apoya un efecto fundador para esta mutación en esta región, lo que pudo haber sido precedido por una separación o cuello de botella (¿socio-cultural o religioso?) de estados premutacionales de la alteración y/o de portadores SCA2 los que con el tiempo fueron siendo más frecuentes (Laffita-Mesa et al., 2008).
Modelaciones bioinformáticas usando marcadores genéticos de 13 familias SCA2 holguineras sitúan el origen más probable de la mutación alrededor de la fundación del Hato Holguinero (Laffita-Mesa et al., 2008; Laffita-Mesa et al., en preparación). Si bien estas predicciones apuntan a un inicio más probable, este proceso pudo extenderse durante 300 años en los que varias olas de inmigrantes, muchos de ellos emparentados, habitaron la región. Es poco probable que individuos enfermos con SCA2 hayan sido los iniciadores, ya que estigmas religiosos y sociales de la época los limitarían para establecer familias, a menos que fueran personas influyentes. También para ellos sería muy difícil hacer largos viajes desde la entonces metrópolis hasta Cuba. En España no existe una población SCA2 con proporciones similares en su frecuencia (Pujana et al., 1999, Mayo-Cabrero et al., 2000, Infante et al., 2005), lo que a su vez brinda poco sustento a la idea de un cromosoma mutante (con penetrancia completa) como evento fundador inicial en Holguín.
El efecto fundador está sustentado, entre otras cosas, por la coexistencia en la misma región y por más de un siglo, de apellidos comunes en las familias SCA2 más numerosas. Existen familias pequeñas (aparentemente de novo) las cuales en su mayoría pueden conectarse con las más grandes. Esto sugiere que, aunque el apellido fundador de las familias SCA2 pudo haberse perdido, quedando evidencias en las tumbas de los cementerios de la región, todas parten de un tronco genético común (Orozco-Diaz et al., 1990). Estas observaciones son respaldadas por estudios de familias SCA2 cubanas y españolas (Laffita-Mesa et al., en preparación), las cuales indican un alto grado de similitud genética entre ellas. Esta similitud, no es particular de estos dos grupos poblacionales, ya que se ha demostrado universalidad de este haplotipo SCA2 (Ramos et al., 2010). La existencia de un haplotipo SCA2 universal apoyaría, por tanto, no solo el efecto fundador antes mencionado sino también un ancestro común y mecanismo genético similar para todas las familias SCA2.
Indirectamente, la existencia de una población fundadora de SCA36 en Costa da Morte, Galicia, así como la alta tasa y larga historia de migración nos sugieren que los movimientos migratorios desde España fueron una fuente importante de introducción de estados premutacionales o de las mutaciones en Latinoamérica y América Central, no solo para la SCA2, sino también para la EH y la fucosidosis, todas concentradas en Holguín (Vázquez-Mojena et al., 2013; Tamayo et al., 2013). A pesar de la mezcla genética que ha tenido lugar en Cuba desde hace más de 500 años, las conexiones ancestrales entre cubanos y españoles (65.05%) es mayoritaria comparada con otros grupos étnicos que emigraron a la isla (Mendizabal et al., 2008). A la vez, Holguín muestra una mayor ascendencia europea comparado con las otras provincias del este de la isla (Marcheco-Teruel et al., 2014; Fortes-Lima et al., 2018).
El tema del efecto fundador de la SCA2, la alta prevalencia en Cuba y la fuente de esta mutación no ha sido agotado del todo, por lo que en el futuro nuevos datos permitirán validar o rechazar las hipótesis actuales. Recientemente se encontró que al igual que Cuba tiene la mayor prevalencia de SCA2, también tiene las mayores frecuencias de alelos largos y existe una correlación entre la frecuencia de estas variantes y la SCA2 (Laffita-Mesa., 2014). Además, hemos podido validar expansiones de novo causales de ELA provenientes de alelos normales largos (Laffita-Mesa et al., 2008; Laffita-Mesa et al., 2013). Por tanto, la existencia de estas variantes largas y/o reversiones puede ser explicativa del origen y sostenimiento de las tasas de prevalencia de SCA2, de aquí el carácter traslacional de entender el origen y diseminación de enfermedades genéticas.
IMPLICACIONES DIRECTAS DE LA INESTABILIDAD DE LA MUTACIÓN SCA2
El riesgo mendeliano a priori para todo portador de la mutación SCA2 es de 50%, lo que subyace al patrón de herencia autosómico dominante de SCA2 (Fig. 4A). Esta es la información básica útil en todo servicio de genética predictiva, la cual deriva del análisis molecular y de la familia (Fig. 4B). En base a la relación edad de inicio vs longitud del segmento CAG se puede estimar cuándo empezarán los síntomas de la enfermedad, por lo que también tiene valor pronóstico para la SCA2. Toda relación genotipo-fenotipo en SCA2 es un fractal de la anterior. Por tanto, los hijos al heredar expansiones mayores que sus padres portadores tendrán pronósticos más desfavorables. La mujer (II-1) que se presenta en la Fig. 4A es una portadora asintomática en la que la mutación se expandió en una unidad de CAG (22/38CAG) y su hijo tiene un riesgo a priori del 50%, el cual es 0% para los descendientes de II-3 con genotipo 22/22CAG. Su hermano II-2 (22/42CAG) al heredar una expansión mayor que ella y que la de su padre ya ha debutado con ataxia.
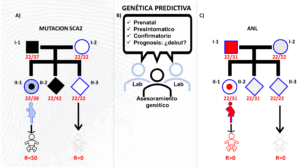
Los alelos largos normales pueden ser estables, expandirse o contraerse. Sea cual sea el escenario, representan un reto más profundo que las mismas mutaciones dado que aun permaneciendo estables pueden combinarse e incrementar el riesgo (R>0) al desarrollo de enfermedades de la motoneurona con mutaciones causales y empeorar el pronóstico (Fig. 4C). Aunque los escenarios presentados son hipotéticos pueden aparecer combinados en la población (Cruz-Mariño et al., 2014).
Los factores genéticos de riesgo se pueden agrupar en aquellos 1) causales o directamente patogénicos, y que determinan un comportamiento mendeliano; 2) los polimorfismos donde la suma o interacción de sus efectos determinarían las enfermedades complejas y 3) aquellos con un efecto mayor y una frecuencia mediana en la población (Manolio et al., 2009) (Fig. 5A). Se ha propuesto este modelo explicativo para la contribución genética en la ELA y se ha validado que alelos largos o intermedios de ATXN2 contribuyen medianamente al fenotipo cuando existe otra mutación como la expansión de hexanucleótidos en C9orf72 (Cady et al., 2014; Dekker et al., 2016) llegándose incluso a estimar la contribución al fenotipo de estos alelos en ELA (Sproviero et al., 2017). También existe la observación de un efecto aditivo de dos alelos de 31 CAG contribuyendo al desarrollo de SCA2 en la ancianidad (Tojima et al., 2018) así como de un paradójico efecto de doble-filo, protector o dañino, en SCA2 (Almaguer-Mederos et al., 2017). Todo esto sugiere que los alelos largos e intermedios complican el diagnóstico predictivo y que falta bastante para entender su rol en las enfermedades asociadas con ATXN2.
TERAPIA ANTISENTIDO PARA SCA2 Y ELA: ¿DOS POR EL PRECIO DE UNO?
Dado que las mismas mutaciones en ATXN2 por una parte causan SCA2 y, por otro lado, una versión más corta de la misma alteración modifica el riesgo para ELA, se está desarrollando una terapia común. Para esto se usan oligonucleótidos antisentido que desencadenan el silenciamiento del ARN defectuoso. Estos son fragmentos de ácidos nucleicos con modificaciones químicas que aumentan su afinidad de unión por ARN mensajero. El mecanismo molecular, al menos en este caso, se fundamenta en la degradación de ARNs defectuosos, que ocurre en la célula como proceso de regulación de la expresión génica.
Aunque existen distintos acercamientos para esta terapia, en los dos estudios preclínicos específicos para ATXN2 se emplearon oligonucleótidos antisentidos (ASO) que silencian ambas versiones del ARN de ATXN2 (Fig. 5B). Scoles y colaboradores rastrearon cientos de moléculas antisentido y una de ellas fue seleccionada porque disminuía significativamente la expresión de ATXN2 in vitro (Scoles et al., 2017). Posteriormente, estudiaron ratones ATXN2-KIN con la mutación SCA2 con un fragmento de 72CAG. A estos animales, que, además, mostraban fenotipo similar a la enfermedad en humanos, se les inyectó vía intracerebroventricular el ASO con afinidad por el ARN de ATXN2. Esto inhibió la traducción de ATXN2, y restituyó el patrón de descargas de potenciales de acción de las neuronas de Purkinje, postergando el debut del fenotipo mórbido y mejorando significativamente los efectos neurodegenerativos.
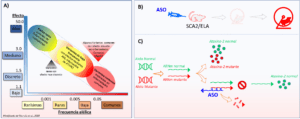
Por su parte, Becker y colaboradores mostraron que este acercamiento extendía la supervivencia de ratones, no con la mutación SCA2, pero sí en un modelo de ELA con mutación en TDP43. También, mostraron que al cruzar ratones KO para la mutación SCA2 con aquellos mutantes de TDP-43 disminuía las patologías asociadas y una mejora significativa del área motriz, y la supervivencia de los animales (Becker et al., 2017). La proteína TDP43 es clave en ELA porque se ha observado que adopta una conformación defectuosa y dañina en esa enfermedad. Además, junto a la ataxina-2 cambia su localización fisiológica a sitios donde ocurre la muerte de las motoneuronas. Esto es debido a la interacción anómala de la ataxina-2 con segmentos de poliQ intermedios y ARNs específicos (Elden et al., 2010).
Estudios del transcriptoma de médula espinal y cerebelo de estos mismos ratones indicaron que hubo una corrección significativa de la expresión de al menos cuatro grupos de genes. Estos estaban encargados de la biosíntesis lipídica, del colesterol, la inmunidad innata y la formación de autofagosomas. Estos últimos son esenciales para el reciclaje de orgánulos celulares deteriorados o aberrantes. De manera general estos hallazgos nos hablan de los procesos celulares que se afectan toda vez que ocurren variantes genéticas en ATXN2 que conlleven a su ausencia o su deposición anormal (Scoles et al., 2020).
Hoy en día existen varios obstáculos por sortear en el uso de ASOs dirigidos a silenciar el ARN mensajero defectuoso de ATXN2. Estos van desde el silenciamiento no alelo específico usado en esta investigación, la administración, biodistribución, el efecto off-target o la citotoxicidad, hasta el hecho de que los ASOs no filtran a través de la barrera hematoencefálica. Sin embargo, la FDA ha aprobado la terapia antisentido para tratar algunas distrofias musculares, y ensayos clínicos fase 3 se están desarrollando para la enfermedad de Huntington y para ELA asociada con variantes en el gen SOD1. Por tanto, en la medida que se desarrolle esta tecnología estos temas de seguridad y eficacia serán aclarados.
Estos hallazgos, aunque preclínicos, son prometedores, y sugieren que la corrección del defecto genético es compatible con la vida, y potencialmente mejoraría la enfermedad humana. Casi veinticinco años después del descubrimiento de la mutación se abre una puerta de esperanza no solo para SCA2, sino además para otras enfermedades estrechamente vinculadas con esta expansión de CAG.
CONSIDERACIONES FINALES
Las alteraciones genéticas en ATXN2 lo convierten en modelo de complejidad genética resumida en un solo gen. El estudio de los mecanismos genéticos del gen ataxin-2 lo convierten, posiblemente en uno de los pocos modelos donde un solo gen provoca varias enfermedades y otro grupo de alteraciones. Primordialmente, el marcado efecto umbral del segmento CAG hace que gradualmente este gen pase de modificador a causal de un amplio espectro de enfermedades que cursan con trastornos de movimiento hasta enfermedades metabólicas. Dentro de este grupo está la SCA2, que muestra alta variabilidad fenotípica como producto de la perdida crónica de las funciones de ataxina-2, empezando mucho antes del debut de signos clínicos más evidentes. Paradójicamente, esta pérdida o ausencia está dada por la ganancia de una función tóxica causada por la expansión de CAG en el exón 1 que a su vez causa deposición y acumulación/agregación anormal de ataxina-2 atascando varios procesos celulares con la consecuente muerte de neuronas. Entender esto al detalle, así como la inestabilidad del segmento de CAG/CAA guiará a la ciencia futura en la mejora de varias enfermedades humanas. Es necesario determinar el momento óptimo antes o después del debut de los síntomas para que estas terapias potenciales sean seguras y efectivas. Además, entender los mecanismos genéticos de ATXN2 permitirá el diseño de ensayos clínicos basados en características individuales y de poblaciones fundadoras. Si entendemos a profundidad SCA2, podremos predecir que otras enfermedades están relacionadas con ATXN2. Basados en el prototipo que es ATXN2, podremos intervenir en otras enfermedades con causas genéticas similares, las cuales, a pesar de su rareza, no merecen la orfandad de atención y tratamientos efectivos.
Declaración de ausencia de conflictos de intereses
Nada a declarar.


