INTRODUCCIÓN
¿Existen enfermedades raras y enfermedades comunes, o únicamente una miríada de expresiones únicas de la enfermedad en los pacientes individuales? El debate entre los “agrupadores” que favorecen las clasificaciones amplias, con características distribuidas en rangos y pocas divisiones, y los “divisores”, que favorecen el reconocimiento de las diferencias de matiz, las características específicas y considerar muchas divisiones, es duradero y posiblemente interminable. No obstante, con el impacto de la secuenciación masiva de exomas humanos, los divisores han ganado considerable terreno en la última década (Conley, 1992; Endersby, 2009; McKusick, 1969; Smith et al., 2022). Esta tendencia ya estaba clara en 2010, cuando se vio que el número de condiciones médicas conocidas se había expandido desde un puñado a casi 5000 en tan solo dos siglos.
Esta situación no debería resultar sorprendente para médicos o biólogos, puesto que los nombres que proporcionamos a las enfermedades son simples etiquetas. El uso de palabras es un frágil intento de describir una percepción unificada transitoria de una realidad biológica altamente heterogénea en evolución. Los pacientes son entidades únicas, idiosincráticas, diferentes, no solo unos de otros sino también de sí mismos en diferentes puntos temporales. Incluso los gemelos idénticos no son idénticos fenotípicamente y las personas mayores son diferentes de los jóvenes que solían ser. El determinismo de la salud y la enfermedad opera en los organismos vivos, cada uno de los cuales difiere de los objetos inertes en que consiste en una única y diversa colección de células con genomas somáticos que evolucionan tanto genética como epigenéticamente en respuesta, y de forma selectiva, al continuamente cambiante ambiente.
Aún así, la mayor parte de los gobiernos y segmentos sustanciales de la academia médica insisten en categorizar e incluso priorizar la investigación médica en lo que ellos denominan “enfermedades comunes” como opuestas a “enfermedades raras” (Chung et al., 2021). Las enfermedades raras se definen típicamente como aquellas condiciones que afectan a menos de 1 de cada 2000 personas (en la Unión Europea) o 1 de cada 1650 personas (en los EE.UU.), siendo las enfermedades comunes aquellas que tienen una frecuencia por encima de estos umbrales arbitrarios. Paradójicamente, hay más enfermedades “raras” que “comunes” y permanece poco claro si el total del número de pacientes con enfermedades “comunes” realmente supera al del número de pacientes con enfermedades “raras”. En el mundo industrializado, esta dicotomía deriva y refuerza un sesgo hacia el estudio de unas pocas enfermedades de las personas mayores, la mayoría de las cuales son “comunes” a costa de las muchas enfermedades de la infancia, la mayoría de las cuales son “raras”.
COVID-19 representa un ejemplo reciente de “enfermedad común”. Aquí revisamos cómo el enigma de la “común” COVID-19, que es esencialmente un problema geriátrico, fue resuelto a nivel molecular y celular a través de la convergencia en 2020 de líneas de investigación pediátrica en dos condiciones genéticas raras que habían permanecido separadas hasta ese momento: los errores innatos de la inmunidad antiviral mediada por IFN de tipo I (variantes en los genes que controlan la inmunidad mediada por IFN de tipo I) y los errores innatos subyacentes a la producción de autoanticuerpos frente a IFN de tipo I (variantes del gen AIRE que controla la tolerancia de las células T).
ERRORES INNATOS DE LA INMUNIDAD MEDIADA POR IFN TIPO I FRENTE A LOS VIRUS
En la actualidad existen 21 errores innatos de la inmunidad mediada por IFN tipo I (Tabla 1 y Figura 1).
Tabla 1. Errores innatos de la inmunidad mediada por IFN tipo 1.
| Gen | Año | Herencia | YFV-17D | MMR/MMRV | HSE | Gripe | COVID-19 | Otros |
| IFNAR1 | 2019 | AR | Sí (Gothe et al., 2022) | Sí (Bastard et al., 2022) | Sí (Bastard et al., 2021) | No | Sí, (Zhang et al., 2020; Abolhassani et al., 2022; Khanmohammadi et al., 2022) | No |
| IFNAR2 | 2015 | AR | Sí (Bastard et al., 2021) | Sí (Duncan et al., 2015; Duncan et al., 2022) | No | No | No | No |
| TYK2 | 2006 | AR | NA | Sí (Zhang et al., 2022; Ogishi et al., 2022) | Sí (Ogishi et al., 2022) | No | Sí, (Zhang et al., 2022; Ogishi et al., 2022) | HSV, VZV, PI3, MC, CMV, RSV, AIV, EBV (Minegishi et al., 2006; Kreins et al., 2015; Ogishi et al., 2022) |
| JAK1 | 2018 | AR | NA | No | No | No | No | No |
| STAT1 | 2003 | AR | NA | Sí (Le Voyer, et al., 2021) | Sí (Dupuis et al., 2003) | Sí (Le Voyer, et al., 2021) | No | PIV, CMV, EBV, HSV, EV, HHV6, RV, VZV, RSV, rotavirus, AIV, MC ((Dupuis et al., 2003; Le Voyer, et al., 202; Chapgier et al., 2006) |
| STAT2 | 2012 | AR | NA | Sí (Hambleton et al., 2013; Alosaimi et al., 2019; Freig et al., 2020; Duncan y Hambleton 2021) | No | Sí (Freij et al., 2020) | Sí (Zhang et al., 2022) | No |
| IRF9 | 2018 | AR | NA | No | No | Sí (Hernandez et al., 2018) | Sí (Levy et al., 2021) | VZV, EV (Bravo Garcia-Morato et al., 2019) |
| IRF7 | 2015 | AR | NA | No | No | Sí (Ciancanelli et al., 2015; Campbell et al., 2022) | Sí (Zhang et al., 2020; Campbell et al., 2022) | RSV, TBE virus, AIV (Campbell et al., 2022) |
| TLR3 | 2007 | AR, AD | NA | No | Sí (Guo et al., 2011; Lim et al., 2014; Hautala et al., 2020; Zhang et al., 2007) | Sí (Schmidt et al., 2021) | Sí (Zhang et al., 2020) | VZV (Liang et al., 2020), EV (Kuo et al., 2022) |
| TRIF/TICAM1 | 2011 | AR, AD | NA | No | Sí (Sancho-Shimizu et al., 2011) | No | No | No |
| UNC9381 | 2006 | AR | NA | No | Sí (Casrouge et al., 2006) | No | No | No |
| TBK1 | 2012 | AR, AD | NA | No | Sí (Herman et al., 2012; Taft et al., 2021) | No | Sí (Schmidt et al., 2021) | No |
| IRF3 | 2015 | AD | NA | No | Sí (Andersen et al., 2015) | No | No | No |
| NEMO | 2005 | XR | NA | Sí, (Miot et al., 2017; Boisson, 2020) | Sí (Audry et al., 2011) | No | No | CMV, HSV, MV, AV (Miot et al., 2017; Boisson, 2020) |
| TLR7 | 2021 | XR | NA | No | No | No | Sí (Zhang et al., 2022; Asano et al., 2021; Abolhassani et al., 2022; Solanich et al., 2021; van der Made et al., 2020) | No |
| MYD88 | 2008 | AR | NA | No | No | No | Sí (García-García et al., en prensa) | No |
| IRAK4 | 2003 | AR | NA | No | No | No | Sí (García-García et al., en prensa) | HHV6 (Nishimura et al., 2021) |
| MDA5 | 2017 | AR, AD | NA | No | No | Sí (Lamborn et al., 2017) | No | RV, EV, RSV (Asgari et al., 2017; Chen et al., 2021) |
| POLR3A | 2017 | AD | NA | No | No | No | No | VZV (Ogunjimi et al., 2017) |
| POLR3C | 2017 | AD | NA | No | No | No | No | VZV (Ogunjimi et al., 2017) |
| MX1 | 2021 | AD | NA | No | No | Sí (H7N9) (Chen et al., 2021) | No | No |
AIV Virus gripe aviar; AV adenovirus; EBV virus Epstein-Barr; EV, enterovirus; CMV citomegalovirus; HHV6, herpesvirus humano 6; HSV virus herpes simple; MC mollusum contagiosum; MV virus del sarampión, PI3 virus parainfluenza 3; PIV virus parainfluenza; RSV virus respiratorio sincitial; RV rinovirus; TBE, encefalitis transmitida por garrapatas, VZV virus zoster varicela; AR autosómica recesiva; AD autosómica dominante; XR ligado al X recesivo.
Errores innatos de ISGF3 (STAT1, STAT2 y IRF9)
El primer error innato de la inmunidad mediada por IFN de tipo I fue reportado en 2003, en un niño con una deficiencia autosómica recesiva completa de STAT1 que presentaba encefalitis por el virus del herpes simple (HSE, herpes simplex virus encephalitis, por sus siglas en inglés) (Dupuis et al., 2003) . El papel de los errores innatos de la inmunidad mediada por IFN de tipo I en la HSE no fue demostrado de forma inequívoca hasta 20 años después cuando se identificó un niño con HSE debida a una deficiencia autosómica recesiva de la cadena 1 del receptor IFN-α/β (IFNAR1) (Bastard et al., 2021). La deficiencia autosómica recesiva completa de STAT1 suprime las respuestas dependientes del factor activador de GAS (GAF-dependiente) y la respuesta dependiente del factor génico 3 estimulado por IFN (ISGF3-dependiente) a los IFN I, II y III, y a IL-27.
En total, en la actualidad se han reportado 24 pacientes con deficiencia autosómica recesiva de STAT1 completa (Le Voyer et al., 2021). Esta condición es el error innato de inmunidad mediada por IFN de tipo I clinicamente más grave, con consecuencias mucho más graves que la deficiencia de STAT1 autosómica recesiva parcial, que se ha reportado en otros ocho pacientes (Le Voyer et al., 2021). La presentación clínica se produce de forma temprana en la vida y la mortalidad es elevada. Predispone a los pacientes a
un amplio rango de enfermedades virales (Tabla 1). Sin embargo, pronto se notó que, paradójicamente, estos pacientes no eran vulnerables a ciertas infecciones virales comunes (Chapgier et al., 2006).
Con deficiencia autosómica recesiva completa de STAT2 solo se han reportado 13 pacientes (Hambleton et al., 2013; Moens et al., 2017; Shahni et al., 2015; Alosaimi et al., 2019; Freij et al., 2020; Meyts et al., 2021; Zhang et al., 2022) y con deficiencia autosómica recesiva completa del factor regulador de IFN 9 (IRF9) dos pacientes (Hernandez et al., 2018; Bravo García-Morato et al., 2019; Levy et al., 2021). La alteración selectiva de las respuestas al IFN I y al IFN III dependiente de ISGF3, con una inmunidad mediada por IFN de tipo I, II y III dependiente de GAF intacta probablemente explica el fenotipo clínico más suave de estos pacientes, que presentan un rango más estrecho de enfermedades virales a nivel global e individual (Tabla 1).
Errores innatos de IFNAR1 y IFNAR2
Las evidencias de que las enfermedades virales de los pacientes con deficiencia de STAT2 y IRF9 resultan en deficiencias en la inmunidad mediada por el IFN de tipo I son proporcionadas por la similitud de sus infecciones virales con las de los pacientes con deficiencia autosómica recesiva de IFNAR1 (Bastard et al., 2021; Hernandez et al., 2019; Gothe et al., 2022; Bastarde et al., 2022; Zhang et al., 2020; Abolhassani et al., 2022; Khanmohammadi et al., 2022) o IFNAR2 (Duncan et al., 2015; Passarelli et al., 2020; Bastard et al., 2021; Duncan et al., 2022). Se han reportado hasta 18 pacientes con deficiencia autosómica recesiva de IFNAR1 y ocho con deficiencia autosómica recesiva de IFNAR2. Estos pacientes son raros globalmente, pero alrededor de uno de cada 1000 individuos con ascendencia de la polinesia occidental o inuit son deficientes para IFNAR1 o IFNAR2, respectivamente, debido a la presencia de alelos nulos en IFNAR1 y INFNAR2 que son sorprendentemente “comunes” (definidos como aquellos con una frecuencia del alelo menor mayor del 1%) en las regiones del Pacífico y Ártico (Bastard et al., 2022; Duncan et al., 2022; Meyts, 2022). Sorprendentemente, solo se han reportado unas pocas enfermedades virales en pacientes con deficiencia de IFNAR1 o IFNAR2 (Tabla 1). Las enfermedades virales más destacadas en estos pacientes antes de la pandemia de COVID-19 han sido la HSE y la gripe crítica. Es destacable que los pacientes son resistentes a la mayoría de los virus comunes. El número de pacientes, su diversidad, el pequeño rango de enfermedades virales, su penetrancia incompleta y la ocurrencia de alelos deletéreos comunes en al menos tres ascendencias convergen en sugerir que los IFN tipo I humanos son esenciales para la defensa del hospedador frente a un pequeño rango de virus. Esta observación sugiere que hay mecanismos de inmunidad antiviral intrínsecos de las células independientes del IFN tipo I, que podrían incluir factores de restricción específicos de tejido y de virus (Nathan, 2021; Palludan et al., 2021; Zhang et al., 2019).
Errores innatos de JAK1 y TIK2
IFNAR1 está asociado constitutivamente con JAK1 e IFNAR2 está asociado constitutivamente con la tirosina quinasa 2 (TYK2) (Meyts y Casanova, 2021) en sus respectivas rutas de señalización y se han reportado pacientes con deficiencias autosómicas recesivas en JAK1 o TYK2.
La deficiencia autosómica recesiva de JAK1 se ha reportado solo de forma parcial en un único paciente que presentaba algunas enfermedades virales debido a su impacto sobre los IFN tipo I (Eletto et al., 2016; Daza-Dajigal et al., 2022). En total se han reportado 40 pacientes con deficiencia autosómica recesiva de TYK2 desde 2006 (Boisson-Dupuis et al., 2018; Fuchs et al., 2016; Kerner et al., 2019; Minegishi et al., 2006; Kreins et al., 2015; Ogishi et al., 2022). Dos de estos pacientes tenían un defecto parcial en rutas de señalización, 25 tenían un defecto completo (con o sin expresión), tres tenían un defecto selectivo raro en la ruta de IL-23 y alrededor de uno de cada 500 individuos de ascendencia europea era homocigoto para la variante P1104A del gen TYK2, que también altera de forma selectiva las respuestas a IL-23. Todos presentaban enfermedad grave por micobacterias debido a respuestas alteradas a IL-23. La respuesta celular al IFN tipo III de estos pacientes con deficiencia para TYK2 autosómica recesiva parece mantenerse y las respuestas a los IFN tipo I solo están afectadas parcialmente y únicamente en pacientes con deficiencia de TYK2 completa o parcial, el 60% de los cuales tuvo enfermedad viral (Ogishi et al., 2022). La señalización residual de IFN tipo I probablemente explica la relativa rareza y la naturaleza benigna de sus enfermedades virales (Tabla 1) (Zhang et al., 2022; Ogishi et al., 2022).
Errores innatos de NEMO, TBK1, IRF3 y IRF7
Los IFN tipo I son inducidos cuando las células son estimuladas o infectadas, con o sin replicación viral, y dependen de una familia de factores de transcripción y reguladores para su producción. Una deficiencia autosómica recesiva de IRF7, un regulador transcripcional clave de los IFN tipo I, se reportó por primera vez en un niño de tres años de edad con neumonía asociada a gripe (Ciancanelli et al., 2015). La deficiencia autosómica recesiva de IRF7 se reportó recientemente en otros seis pacientes de cinco familias (Campbell et al., 2022). Curiosamente, el fenotipo de infección viral de estos pacientes estaba restringido al tracto respiratorio (Tabla 1). Es posible que los niveles residuales de IFN-β sean responsables del mejor control de los virus en estos pacientes que en los pacientes con deficiencia de IFNAR1 o IFNAR2, a pesar de la falta de inducción de IFN tipo I y tipo III dependiente de IRF-7. La inmunidad adaptativa a los virus podría compensar los defectos de la inmunidad mediada por IFN tipo I en estos pacientes (Campbell et al., 2022). Por el contrario, se ha reportado un niño con una forma autosómica dominante parcial de deficiencia de IRF que tenía HSE (Andersen et al., 2015).
Un defecto en un punto superior de la cascada de inducción de IFN tipo I, la deficiencia autosómica dominante de kinasa 1 de unión a TANK (TBK1), también implica HSE (Herman et al., 2012). Paradójicamente, se encontró que la deficiencia autosómica recesiva de TBK1 implicaba una condición autoinflamatoria en cuatro pacientes de edades comprendidas entre los 7 y los 32 años sin historia de enfermedad viral grave (Taft et al., 2021). Finalmente, también se ha reportado un niño con HSE y una variante específica de NEMO, que codifica para un componente regulador del complejo IKK en la ruta canónica NF-κB (Audry et al., 2011; Casanova et al., 2014). El mecanismo probablemente implica la alteración de la inducción del IFN tipo I, posiblemente a través de su impacto sobre IFN-β.
Errores innatos de TLR3, TRIF, UNC93B, MDA5 y POLR3A/C
La activación de la producción del IFN tipo I depende con frecuencia de los receptores de detección viral y sus reguladores. Los defectos autosómicos recesivos de TLR3, TRIF/TICAM1 o UNC93B subyacen a la HSE del cerebro anterior (Tabla 1) (Casrouge et al., 2006; Guo et al., 2011; Lim et al., 2014; Sancho-Shimizu et al., 2011) con una penetrancia mayor a la de las correspondientes formas autosómicas dominantes de deficiencia en TLR3 y TRIF (Casrouge et al., 2006; Lim et al., 2014; Sancho-Shimizu et al., 2011; Liang et al., 2020). TLR3 es un sensor endosomal de ARN de doble cadena (ARNdc) que se genera como intermediario o subproducto de la infección viral. También controla los niveles tónicos basales del IFN tipo I en fibroblastos y neuronas corticales, y posiblemente también en células epiteliales respiratorias, con la posible participación de agonistas endógenos hasta ahora desconocidos (Gao et al., 2021). TRIF/TICAM1 es un adaptador y UNC93B es un componente de unión en la vía secretora. TRIF se une casi exclusivamente a TLR3 (y TLR4), mientras que UNC93B también es necesario para las respuestas a los otros tres sensores endosomales de ácidos nucleicos, TLR7, TLR8 y TLR9.
La deficiencia de MDA5 autosómica recesiva es menos rara en la población general, ya que al menos un alelo de pérdida de función tiene una frecuencia de casi el 1%. Sin embargo, sólo se han descrito cuatro pacientes con esta deficiencia (Asgari et al., 2017; Chen et al., 2021; Lamborn et al., 2017): tres de estos pacientes no emparentados presentaban enfermedades víricas respiratorias distintas de la gripe (Asgari et al., 2017; Chen et al., 2021; Lamborn et al., 2017) y el cuarto presentaba encefalitis por enterovirus del tronco encefálico (Chen et al., 2021). TLR3 detecta ARN de cadena doble en los endosomas, mientras que MDA5 detecta ARN de cadena doble en el citosol. Por último, se han descrito variantes de los genes que codifican las subunidades A y C del sensor de ADN de cadena doble de la ARN polimerasa III (POLR3A y POLR3C) en pacientes con encefalitis por el virus de la varicela zóster (Ogunjimi et al., 2017).
Errores innatos de TLR7, IRAK4 y MyD88
Otros errores innatos de la inmunidad IFN tipo I afectan a la detección de ARN monocatenario en lugar de ARN de doble cadena. Casi todos los pacientes con deficiencia IRAK4 o MyD88 autosómica recesiva descritos entre 2003 y 2019 presentaban infecciones bacterianas piógenas, pero no infecciones víricas (Picard et al., 2003; von Bernuth 2008). Dos excepciones fueron pacientes con encefalitis por herpesvirus humano 6 (Nishimura et al., 2021; Tepe et al., 2022). Esto llevó a sugerir que los TLR7, TLR8 y TLR9 humanos, que son sensores endosomales de ácidos nucleicos y dependen todos de IRAK4 y MyD88 para su señalización, eran redundantes para la defensa del huésped contra la mayoría de los virus actuales y comunes (Picard et al., 2003; von Bernuth et al., 2008). Además, se descubrió que los pacientes con deficiencia autosómica recesiva de UNC93B, cuyas células no pueden responder a TLR3, TLR7, TLR8 y TLR9, eran propensos a HSE, al igual que los pacientes con deficiencia de TLR3 (Casrouge et al., 2006). Esto también sugería que TLR7, TLR8 y TLR9 eran en gran medida redundantes en la defensa del huésped (Casrouge et al., 2006). Esta idea resultaba paradójica, debido a que los genes que codifican los cuatro TLR endosomales sensores de ácidos nucleicos, incluido TLR3, estaban sometidos a una selección negativa más fuerte que la aquellos que codifican los otros TLR (Quach et al., 2013). Como se detalla más adelante, este enigma se resolvió cuando se descubrió que la deficiencia recesiva de TLR7 ligada al cromosoma X era una etiología genética de la neumonía crítica COVID-19 (Asano et al., 2021). Posteriormente se descubrió que los pacientes con deficiencia de IRAK4 o MyD88 presentaban un riesgo muy elevado de neumonía COVID-19 potencialmente mortal (García-García et al., 2023).
Estos hallazgos son coherentes con la demostración de que las células dendríticas plasmacitoides (PDC, por sus siglas en inglés) dependen de IRAK4 y TLR7 para la detección del SARS-CoV-2 y la producción de IFN tipo I (Asano et al., 2021, Onodi et al., 2021) y con la observación de que los pacientes con leucemia linfocítica crónica tienen recuentos disminuidos de pDC y son propensos a la neumonía hipoxémica por COVID-19 (Smith et al., 2022).
Errores innatos de MX1
El primer error innato humano de un gen estimulado por IFN (ISG, IFN stimulated gene) que se describió fue la deficiencia autosómica recesiva de ISG15 (Bogunovic et al., 2012). Los pacientes afectados no presentaban enfermedad vírica, e incluso se observó que sus células eran inusualmente resistentes a la infección vírica (Zhang et al., 2015). Estas células tienen niveles anormalmente altos de actividad IFN tipo I in vivo, y los pacientes presentan una interferonopatía tipo I que se manifiesta con calcificaciones cerebrales (Zhang et al., 2015). El mecanismo subyacente implica una regulación no controlada de la ruta molecular de respuesta al IFN tipo I dependiente de USP18 y STAT2, como confirma la identificación de pacientes homocigotos para variantes de STAT2 que alteran la interacción de STAT2 con USP18 (Duncan et al., 2019; Gruber et al., 2020; Duncan y Hambleton, 2021) y de pacientes con deficiencia autosómica recesiva completa o parcial de USP18 (Martin-Fernandez et al, 2022; Meuwissen et al., 2016; Alsohime et al., 2020), que también presentan una interferonopatía tipo I. Paradójicamente, los dos primeros trastornos recesivos de genes estimulados por IFN (las deficiencias de ISG15 y USP18) implican una interferonopatía tipo I que puede aumentar potencialmente la resistencia a los virus.
Hasta 2021 no se informó de una forma autosómica dominante de deficiencia de MX1 en pacientes chinos con enfermedad crítica debida al virus de la gripe aviar (Chen et al., 2021). La GTPasa MX1 inducida por IFN se identificó por primera vez en 1986 (mediante estudios de complementación) como esencial para la inmunidad frente al virus de la gripe en diversas cepas de ratón (Staeheli et al., 1986). Este descubrimiento seminal fue el punto de partida para la búsqueda de genes de susceptibilidad para la infección (Casnova y Abel, 2022). Treinta y cinco años después se descubrió un enriquecimiento en variantes germinales raras en MX1 en pacientes chinos con gripe aviar grave (Chen et al., 2021). La mayoría de estas variantes de pérdida de función también son dominantes negativas.
ERRORES CONGÉNITOS DE TOLERANCIA AL IFN DE TIPO I
APS1: características clínicas e historia
Una línea de investigación independiente condujo al descubrimiento de autoanticuerpos contra los IFN tipo I que alteran su actividad (Tabla 2 y Figura 1). La mayoría, si no todos, los pacientes con síndrome poliglandular autoinmune tipo 1 (APS1; OMIM #240300), también conocido como distrofia ectodérmica poliendocrinopatía autoinmune (APECED), desarrollan un defecto de la inmunidad IFN tipo I a través de una respuesta autoinmune adquirida frente a los IFN tipo I (Ahonen et al., 1990).
Tabla 2. Errores innatos de la tolerancia al IFN tipo 1.
| Gen | Año | Herencia | YFV-17D | MMR/MMRV | HSE | Gripe | COVID-19 | Otros |
| AIRE | 1998 | AR, AD | NA | No | No | No | Sí (Bastarde et al., 2021; Bastard et al., 2020; Beccuti et al., 2020, Meisel et al., 2021; Schidlowski et al., 2022) | VZV (Hetemaki et al., 2021) |
| NIK | 2017 | AR | NA | No | No | No | Sí (Le Voyer et al., 2023) | No |
| NFKB2 | 2013 | AD | NA | No | No | Si | Sí (Le Voyer et al., 2023) | VZV (Le Voyer et al., 2023) |
| RELB | 2015 | AR | NA | No | No | No | Sí (Le Voyer et al., 2023) | VZV (Le Voyer et al., 2023) |
| FOXP3 | 2000 | XR | NA | No | No | No | NA | No |
| RAG1 y RAG2 | 1998 | AR | NA | Sí | No | sí (Le Voyer et al., 2023) | NA | VZV, CMV, RV, RSV (Walter et al., 2015) |
CMV citomegalovirus; RSV virus respiratorio sincitial; RV rinovirus; VZV virus zoster varicela; AR autosómica recesiva; AD autosómica dominante; XR ligado al X recesivo.
La APS1 se describió clínicamente por primera vez en 1943 (Sutphin et al., 1943). Se caracteriza por el desarrollo de múltiples enfermedades autoinmunes específicas de órgano en un solo paciente y su herencia es típicamente autosómica recesiva. Es globalmente rara (1 de cada 200.000), pero con una prevalencia al menos 10 veces mayor en Escandinavia (1 de cada 14.000), debido a efectos fundadores (Constantine y Lionakis, 2019). Las características autoinmunes varían entre pacientes individuales, pero las características clínicas más comunes son la enfermedad de Addison, el hipoparatiroidismo y una susceptibilidad inusualmente selectiva a la candidiasis mucocutánea crónica (CMC). Esta tríada central se observa en aproximadamente el 75% de los pacientes. Incluso dentro de una misma familia, las afecciones autoinmunes que se desarrollan pueden diferir entre los parientes afectados.
El tratamiento de los pacientes con APS1 suele incluir cuidados de apoyo y, con frecuencia, terapia de sustitución de los órganos afectados, con inmunosupresión utilizada ocasionalmente para tratar las características más graves, como la hepatitis autoinmune (Perheentupa, 2006). El resultado clínico general de los pacientes con APS1 es muy variable, pero la mortalidad alcanza el 50% a la edad de 45 años, normalmente debido al efecto acumulativo de múltiples características de la enfermedad y sus secuelas (Borchers et al., 2020).
El descubrimiento que vincula AIRE con APS1
Dado el patrón típico de herencia autosómica recesiva para APS1, las aproximaciones de ligamiento físico mapearon el gen defectuoso en el cromosoma humano 21 en 1994 (Aaltonen et al., 1994). Continuando con esta laboriosa aproximación de ligamiento, dos grupos reportaron de forma simultánea la identificación del gen defectuoso en 1997. Se acordó entonces llamar al gen “regulador autoinmune” (AIRE), dado el fenotipo clínico de los pacientes con APS1 (Aaltonen et al., 1997; Nagamine et al., 1997). El nuevo gen no mostraba una marcada similitud de secuencia con ningún gen conocido y se pensó que codificaba una proteína de 545 aminoácidos con al menos cuatro dominios distintos. El análisis de la secuencia del gen AIRE indicó que contenía un dominio de localización nuclear (Aaltonen et al., 1997; Nagamine et al., 1997). Además, la tinción para la proteína dio como resultado un patrón nuclear moteado en células que expresaban activamente el gen (Heino et al., 1999).
La búsqueda de este gen allanó el camino hacia el descubrimiento de un regulador crítico de la tolerancia inmunitaria, debido a que los pacientes con esta enfermedad eran portadores de variantes que se predecía que serían de pérdida de función (por ejemplo, variantes sin sentido) cuando estuvieran en homocigosis. Hasta 2014 no se descubrió que variantes heterocigotas y dominantes negativas de AIRE subyacen a una forma autosómica dominante de APS1, tanto en familias con múltiples afectados como en casos esporádicos (Oftedal et al., 2015; Cetani et al., 2001; Su et al., 2008), típicamente con un fenotipo más leve.
Las principales pistas sobre la función de AIRE las proporcionaron inicialmente los estudios que asignaron su expresión al timo y, en particular, a las células epiteliales de la médula tímica (mTEC, por sus siglas en inglés) (Anderson et al., 2002). Se desarrolló un modelo de ratón knockout que también presentaba múltiples afecciones autoinmunes (Anderson et al., 2002) cuyo análisis detallado de las mTEC dio lugar a un modelo en el que AIRE promueve la expresión de una amplia gama (miles) de autoantígenos específicos de tejido (TSA, por sus siglas en inglés), todos ellos expresados en los tejidos aislados (Anderson et al., 2002, Sansom et al., 2014).
Otra interesante imagen de expresión génica está siendo descifrada en las mTEC, una fracción de las cuales se diferencia aún más tras la expresión de AIRE y adquiere un programa de expresión génica que refleja algunos tejidos periféricos como el enteroendocrino, el epitelio respiratorio, el epitelio cutáneo maduro y las células en penacho, que también contribuyen a la expresión de TSA (Michelson et al., 2022; Bautista et al., 2021; Miller et al., 2018; Bornstein et al., 2018).
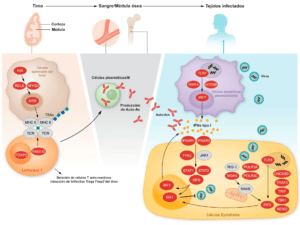
Durante el desarrollo, los timocitos transitan por el compartimento medular, donde estas células sufren un paso crítico de selección negativa en el que las células T autorreactivas son eliminadas por el reconocimiento de autoantígenos en la médula. AIRE controla la tolerancia inmunitaria de las células T impulsando la expresión de “lo propio” en la médula, de modo que las células T autorreactivas que se desarrollan por casualidad pueden reconocer “lo propio” y ser eliminadas del conjunto de células T en desarrollo (Figura 1). AIRE también puede promover el desarrollo de una fracción de linfocitos T reguladores CD4+ Foxp3+ protectores frente a la autoinmunidad en la periferia (Malchow et al., 2013). Los estudios en ratones han establecido claramente que este elegante proceso de selección en el timo es notablemente eficaz y con frecuencia da lugar a la eliminación de células T específicas de tejido (Taniguchi et al., 2012). Curiosamente, recientes estudios con células individuales del timo humano también han demostrado que los TSA se expresan en células que expresan AIRE dentro del timo y que estas células expresan con frecuencia las dianas de la respuesta autoinmune en pacientes con APS1 (Bautista et al., 2021).
Mecanismos de las enfermedades endocrinas autoinmunes y fúngicas
El mecanismo central de la enfermedad en los pacientes con APS1 está impulsado por las células T, pero el daño tisular se asocia a menudo con autoanticuerpos específicos del tejido. La enfermedad endocrina en pacientes con APS1 se debe principalmente a una destrucción del órgano afectado mediada por células T y los modelos de ratón han apoyado una respuesta prominente de tipo Th1 en los tejidos afectados (Devoss et al., 2008). Los pacientes con APS1 pueden desarrollar una amplia gama de respuestas autoinmunes, pero, inesperadamente, se ha descubierto que algunas de estas respuestas están dirigidas contra las citocinas.
Como se ha mencionado anteriormente, la candidiasis mucocutánea crónica (CMC) es una afección intrigante prominente en los pacientes con APS1. La CMC se desarrolla en estos pacientes debido a una respuesta autoinmune dirigida contra citocinas Th17 cruciales, como la IL17A y la IL17F, detectándose autoanticuerpos neutralizantes de ambas citocinas en la mayoría de los pacientes (Puel et al., 2010, Kisand et al., 2010). Esta conexión autoinmune se ve reforzada por datos que demuestran que la candidiasis se desarrolla con frecuencia en pacientes con variantes de la línea germinal que afectan a la IL-17A/F y a su receptor IL17RA/RC (Puel et al., 2011) y en pacientes tratados con anticuerpos bloqueantes contra estas citocinas para afecciones inflamatorias (Saunte et al., 2017). Estos hallazgos sugieren que la mayoría, si no todas, las características clínicas de los pacientes con APS1, incluida su característica infección fúngica aislada, son de naturaleza autoinmune.
Autoanticuerpos “silenciosos” contra IFNs tipo I
A partir de 2006 se ha reportado que más del 90% de los pacientes con APS1 desarrollan autoanticuerpos contra los IFN tipo I (Meager et al., 2006). También se han encontrado en pacientes con miastenia gravis, timoma y lupus eritematoso sistémico, y en individuos tratados con IFN-α2 o IFN-β (Mogensen et al., 1981; Meager et al., 1997; Suit et al., 1983; Vallbracht
et al., 1981). El significado clínico de estos autoanticuerpos en general, y en pacientes con APS1 en particular, seguía siendo desconocido, ya que los pacientes con estos autoanticuerpos no mostraban una susceptibilidad consistente a las infecciones víricas.
Los autoanticuerpos contra IFNs tipo I observados en individuos con APS1 están dirigidos casi exclusivamente contra las 13 formas de IFN-α y la única forma ω, raramente contra IFN-β, y aparentemente no contra ε y κ (Meager et al., 2006). Este patrón se identificó como una posible razón de la falta de asociación manifiesta de estos anticuerpos con un fenotipo de susceptibilidad viral. El IFN-β, en particular, es el primer IFN tipo I inducido por los virus en la mayoría de las células.
Como se detalla más adelante, no fue hasta 2020 cuando se descubrió que los pacientes con APS1 presentaban un riesgo muy elevado de padecer neumonía COVID-19 crítica e incluso otras enfermedades víricas (Bastard et al., 2021; Hetemaki et al., 2021). La alta prevalencia de autoanticuerpos contra IFNs tipo I en pacientes con APS1 y timoma sugiere que los defectos de la función del timo pueden desencadenar esta respuesta autoinmune específica. En apoyo de esta noción, se ha demostrado que la expresión de AIRE está alterada en el timoma, conectando el mecanismo de autoinmunidad en pacientes con APS1 hereditario con el timoma adquirido (Cheng et al., 2010).
Otras etiologías en las células epiteliales de la médula tímica (mTEC) de autoanticuerpos contra IFNs tipo I
La expresión de AIRE en mTEC de ratones está impulsada por RANK a través de la vía alternativa NF-κB (Figura 1) (Cheng et al., 2010; Rossi et al., 2007; Akiyama et al., 2008; Hikosaka et al., 2008). De forma consistente, se han encontrado anticuerpos contra IFNs tipo I en pacientes con deficiencias autosómicas recesivas de NIK o RELB y en pacientes con una forma específica de deficiencia autosómica dominante de NF-κB2 debida a variantes C-terminales que impiden la escisión de p100 en p52, resultando en una pérdida de actividad de p52 pero una ganancia de función inhibitoria para p100 (Ramakrishnan et al., 2018). Además, se ha observado que la expresión de AIRE en el timo está alterada en los pacientes estudiados con variantes de RELB o NFKB2. Las deficiencias de la vía alternativa NF-κB pueden, por tanto, subyacer a la producción de autoanticuerpos contra IFNs tipo I a través de una alteración de la expresión de AIRE en las mTECs. Por el contrario, los pacientes con errores innatos de la inmunidad canónica NF-κB analizados no presentaban autoanticuerpos contra IFN tipo I.
Sin embargo, la mayoría de las mujeres con incontinencia pigmentaria debida a heterocigosidad para variantes de pérdida de función de NEMO sí presentan dichos autoanticuerpos, posiblemente debido a la apoptosis de mTECs que expresan el alelo NEMO mutado durante el desarrollo tímico (Bastard et al., 2020). En conjunto, estos hallazgos sugieren que las disfunciones tímicas dependientes de AIRE (variantes deletéreas de AIRE o de los genes que codifican componentes de la ruta molecular inductora de AIRE en las mTEC, o localmente, dentro de un timoma) pueden subyacer a la producción de anticuerpos contra IFNs tipo I.
Otras etiologías de células T de autoanticuerpos contra IFNs tipo I
También se ha descubierto que varios errores innatos intrínsecos de las células T subyacen a los autoanticuerpos contra los IFN tipo I. Los pacientes varones con variantes deletéreas del gen FOXP3 ligado al cromosoma X, que muestran una pérdida de células T reguladoras funcionales, a menudo son portadores de autoanticuerpos contra IFNs tipo I (Rosenberg et al., 2018). Estos pacientes presentan una enfermedad conocida como inmunodesregulación-poliendocrinopatía-enteropatía ligada al cromosoma X (IPEX) (Chatila et al., 2000; Bennet et al., 2001), que tiene características autoinmunes y clínicas que se solapan en parte con las del APS1 (Bennet et al., 2001b). No se ha descrito que presenten enfermedad vírica grave, al menos antes de la terapia inmunosupresora para el trasplante de células madre hematopoyéticas.
Los pacientes con variantes deletéreas en RAG1 o RAG2 e inmunodeficiencia combinada también podrían producir autoanticuerpos contra IFNs tipo I (Walter et al., 2015). Estos pacientes presentan con frecuencia enfermedades por herpes virus, debido a la sola presencia de estos autoanticuerpos contra los IFN tipo I o junto con la deficiencia combinada de células T y B. Las etiologías conocidas de autoanticuerpos contra IFN tipo I afectan, por tanto, a la tolerancia de células T, de forma intrínseca a las células T (RAG, FOXP3) o a través de las mTECs (AIRE y la vía que la induce). Los defectos de AIRE están relacionados con el deterioro de la correcta selección de células T reguladoras (Malchow et al., 2013), y los defectos de RAG1 y RAG2 están relacionados con el deterioro de la expresión de AIRE (Cavadini et al., 2005). En conjunto, estos datos vuelven a relacionar la generación de autoanticuerpos contra IFNs tipo I con la selección en el timo.
NEUMONÍA CRÍTICA POR COVID-19 Y DEFICIENCIA DE IFN TIPO I
El problema, la hipótesis y el enfoque
El problema clave planteado por COVID-19 en 2020 es común a todos los patógenos humanos: ¿qué impulsa la gran variabilidad clínica interindividual observada durante la infección (Casanova y Abel, 2022; Casanova y Abel, 2021)? La tasa global de letalidad por infección (IFR) de COVID-19 en individuos no vacunados fue de alrededor del 1% en todas las edades y sexos. Se observó que el riesgo de muerte se duplicaba cada 5 años de edad, desde la infancia en adelante, siendo el riesgo de muerte 10.000 veces mayor a los 85 años que a los 5 años (Zhang et al., 2022).
Nos planteamos la hipótesis de que la neumonía crítica por COVID-19 podría ser el resultado de errores innatos de la inmunidad en un solo gen, al menos en algunos pacientes (Casanova et al., 2020). La identificación de un error innato causal, incluso en un solo paciente, podría ser suficiente para tirar del hilo mecanicista y revelar otras causas que alteren los mismos mecanismos fisiológicos en otros pacientes (Casanova y Abel, 2022). El COVID Human Genetic Effort (www.covidhge.com) se creó para seguir este enfoque y reclutar al mayor número posible de pacientes en todo el mundo, de modo que pudieran detectarse incluso niveles bajos de homogeneidad genética (Casanova et al., 2020). Los fenotipos y genotipos de los pacientes se pusieron a disposición de todos los equipos del consorcio, lo que facilitó la investigación coordinada y sinérgica de los determinantes genéticos e inmunológicos humanos de la COVID-19 crítica.
Errores innatos de inmunidad frente a la gripe y genes candidatos
La primera hipótesis que se analizó fue que la neumonía crítica debida al virus de la gripe estacional y la neumonía crítica debida al SARS-CoV-2 podrían ser alélicas.
Los pacientes con deficiencia autosómica recesiva de IRF7, deficiencia autosómica recesiva de IRF9 y deficiencia autosómica recesiva o dominante de TLR3 eran propensos a la gripe grave (Lim et al., 2019). Se consideraron otros diez genes, que codifican productos conectados bioquímica e inmunológicamente con los tres genes centrales de susceptibilidad a la gripe y variantes de la línea germinal que ya se había demostrado que subyacen a otras enfermedades víricas graves (Figura 1). Entre los genes considerados se incluyeron los que codifican STAT1 y STAT2, que pronto se confirmaron como genes de susceptibilidad a la gripe (Le Voyer et al., 2021; Freij et al., 2020).
En 23 pacientes con neumonía crítica por COVID-19 se encontraron variantes raras y deletéreas de 8 de los 13 genes candidatos. Once pacientes tenían trastornos dominantes conocidos, mientras que ocho tenían trastornos dominantes potencialmente nuevos. Estos hallazgos se replicaron en una cohorte mayor (Matuozzo et al., 2022).
Las variantes de la línea germinal que afectan a la vía TLR3 sugirieron que los niveles tónicos de IFN tipo I en las células epiteliales respiratorias desempeñan un papel importante en la defensa del huésped frente al SARS-CoV-2 (Gao et al., 2021). Cuatro pacientes con
defectos autosómicos recesivos proporcionaron una visión única de la patogénesis de COVID-19. Dos adultos no emparentados tenían deficiencia autosómica recesiva de IFNAR1, mientras que otros dos tenían deficiencia autosómica recesiva de IRF7 (Zhang et al., 2020). Posteriormente se reportaron otros pacientes con COVID-19 crítica debida a deficiencia autosómica recesiva de IFNAR1 (Zhang et al., 2022; Abolhassani et al., 2022; Khanmohammadi et al., 2022) o de IRF7 (Campbell et al. 2022), así como de un paciente con deficiencia autosómica recesiva de TBK1 (Schmidt et al., 2021). Sorprendentemente, los adultos jóvenes e incluso de mediana edad identificados con déficits autosómicos tan profundos habían permanecido bien hasta que desarrollaron COVID-19.
Búsqueda en todo el genoma: TLR7 e IFN tipo I de nuevo
Una prueba sobre en el cromosoma X encontró un enriquecimiento en variantes raras no sinónimas en un único locus que codifica el sensor de ARN endosomal TLR7 (Asano et al., 2021). La falta de enriquecimiento en el locus TLR8 ligado al cromosoma X sugería no sólo que la mayoría de las variantes de TLR7 eran deletéreas y patogénicas, sino también que el mecanismo de la enfermedad implicaba una alteración de la inducción de IFN tipo I dependiente de TLR7 por las células dendríticas plasmacitoides (PDCs) .
De hecho, TLR7 y TLR8 son ambos sensores endosomales de ARNs solapantes, y ambos señalizan a través de la vía de señalización dependiente de MyD88 e IRAK4, que ya había demostrado ser esencial para la detección del SARS-CoV-2 en las PDCs. Sin embargo, TLR7 se expresa en las PDCs, mientras que TLR8 no (Onodi et al., 2021). Otros experimentos mostraron que la mayoría de las variantes de TLR7 en pacientes con COVID-19 crítico, pero ninguna de las de individuos con infección leve, eran de pérdida de función. La penetrancia fue incompleta entre los familiares de los casos índice.
Las PDCs deficientes en TLR7 tenían respuestas muy alteradas al SARS-CoV-2 (Asano et al., 2021). La deficiencia recesiva de TLR7 ligada al cromosoma X se encontró en alrededor del 1% al 2% de los pacientes varones con COVID-19 crítica. La proporción de adultos con neumonía crítica debida a estos 14 errores innatos, incluidos los defectos autosómicos, fue de alrededor del 3% al 5%, mientras que alrededor del 10% de los niños con neumonía por COVID-19 presentaban deficiencias recesivas no sólo de TLR7 e IRF7, sino también de STAT2 y TYK2 (Zhang et al., 2022).
Una aproximación genómica no sesgada volvió a implicar defectos de la inmunidad mediada por IFN tipo I. Las variantes de la vía TLR3 habían implicado a las células epiteliales respiratorias residentes, pero las variantes de TLR7 implicaron a las PDCs circulantes, indicando que el reclutamiento de estas células en el tracto respiratorio durante la infección por SARS-CoV-2 era esencial para la inmunidad protectora mediada por IFN tipo I.
Pacientes con APS-1 y neumonía hipoxémica por COVID-19
Al principio de la pandemia de COVID-19, varios pacientes con APS1 desarrollaron neumonía crítica por COVID-19 (Bastard et al., 2020; Beccuti et al., 2020). Partiendo de este conocimiento y de la identificación de errores innatos de IFN tipo I en otros pacientes con COVID-19 crítica, se desarrolló una hipótesis unificadora según la cual la susceptibilidad a la neumonía por COVID-19 crítica de los pacientes con APS1 se debía a sus autoanticuerpos preexistentes contra el IFN tipo I.
En una serie internacional de 22 pacientes con APS1 de edades comprendidas entre los 8 y los 48 años, el 86% presentó neumonía hipoxémica, incluido un 68% con enfermedad grave y un 18% que falleció (Bastard et al., 2021). Un estudio más pequeño y unicéntrico de cuatro pacientes confirmó que no todos los pacientes con APS1 infectados por SARS-CoV-2 desarrollaron neumonía hipoxémica (Meisel et al., 2021), mientras que un estudio más reciente informó de varios otros pacientes con APS1 con COVID-19 crítico (Schidlowski et al., 2022). Es importante destacar que los autoanticuerpos contra IFNs tipo I estaban presentes en los pacientes APS1 antes de su infección con SARS-CoV-2 y el desarrollo de la neumonía en COVID-19.
Dado que se ha demostrado que los errores innatos de la inmunidad IFN de tipo I son causantes de neumonía crítica, estos hallazgos proporcionaron una prueba de principio de que los autoanticuerpos que neutralizan el IFN tipo I también pueden ser causantes de neumonía crítica. Por lo tanto, esta enfermedad rara proporcionó una visión clave de uno de los posibles mecanismos subyacentes al desarrollo del curso grave de COVID-19 en algunos sujetos.
Autoanticuerpos contra IFNs tipo I en pacientes con COVID-19 crítica
Sorprendentemente, alrededor del 10% de los pacientes con COVID-19 crítica portaban autoanticuerpos circulantes que neutralizaban altas concentraciones de IFN-α y/o IFN-ω (Bastard et al., 2020). Posteriormente, se comprobó que esta proporción era superior (15%) si se consideraban los pacientes cuyo plasma neutralizaba concentraciones inferiores (Bastard et al., 2021). Rara vez se encontraron autoanticuerpos neutralizantes del IFN-β y los pacientes con autoanticuerpos contra IFNs tipo I representaron colectivamente el 20% de las muertes en todos los grupos de edad y el 20% de los casos críticos entre los pacientes mayores de 70 años.
El riesgo de enfermedad crítica aumentaba tanto con el número de formas de IFN tipo I como con la concentración de IFN neutralizado (Bastarde et al., 2021, Manry et al., 2022). Estos hallazgos se han replicado en 29 poblaciones independientes de todo el mundo (Bastard et al., 2021; Meisel et al., 2021; Schidlowski et al., 2022; Abers et al., 2021; Acosta-Ampudia et al., 2021; Akbil et al., 2022; Busnadiego et al., 2022; Carapito et al., 2022; Chang et al., 2021; Chauvineau-Grenier et al., 2022; Credle et al., 2022; Eto et al., 2022; Frasca et al., 2022; Goncalves et al., 2021; Konig et al., 2021; Lamacchia et al., 2022; Lemarquis et al., 2021; Mathian et al., 2022; Petrikov et al., 2022; Raadsen et al., 2022; Savvaeeva et al., 2021; Simula et al., 2022; Solanich et al., 2021; Soltani-Zangbar et al., 2022; Troya et al., 2021; van der Wijst et al., 2021; Vazquez et al., 2021; Wang et al., 2021; Ziegler et al., 2021). También se ha demostrado que estos autoanticuerpos subyacen a una respuesta retardada del gen estimulado por IFN tipo I en los leucocitos, como lo demuestra la secuenciación de ARN en células individuales (van der Wijst et al., 2021), y en la mucosa nasal, como lo demuestra la secuenciación de ARN (Lopez et al., 2021).
Estos autoanticuerpos se detectaron en muestras de sangre extraídas muy pronto durante la hospitalización (van der Wijst et al., 2021) e incluso en muestras pre-COVID-19 en el pequeño número de pacientes para los que dichas muestras estaban disponibles. Sus niveles en sangre pueden aumentar durante la COVID-19 (Akbil et al., 2022; Shaw et al., 2021). Su prevalencia, estudiada en 33.000 individuos de entre 20 y 100 años de los que se disponía de muestras recogidas antes de 2019 (Bastard et al., 2021) se mantenía estable hasta los 65 años, entre el 0,3% y el 1% en función de las concentraciones neutralizadas, aumentando posteriormente hasta alcanzar el 4% y el 7%, respectivamente, después de los 80 años (Bastard et al., 2021). La prevalencia de autoanticuerpos neutralizantes del IFN-β permaneció estable, en torno al 0,2%, en todos los grupos de edad. Estos hallazgos sugieren que la tasa global de letalidad por infección es mucho mayor en individuos con autoanticuerpos que en aquellos sin estos anticuerpos (Manry et al., 2022).
Desarrollo de una prueba clínica: estratificación del riesgo y enfoques terapéuticos
La presencia de estos autoanticuerpos es el segundo factor de riesgo común más importante para la COVID-19 crítica después de la edad. Si se combinan el riesgo asociado a la edad y el riesgo asociado a la presencia de autoanticuerpos contra IFNs tipo I, la tasa de mortalidad efectiva por COVID-19 puede alcanzar niveles muy superiores al 50% en sujetos mayores de 80 años portadores de autoanticuerpos contra IFNs tipo I (Manry et al., 2022). Esto proporciona un argumento de peso para realizar pruebas de detección de estos anticuerpos en la evaluación inicial de los pacientes diagnosticados de COVID-19, especialmente, aunque no de forma exclusiva, en aquellos que no han sido vacunados.
Recientemente descubrimos que alrededor del 20% de los casos de neumonía hipoxémica «irruptiva» se debían a autoanticuerpos que neutralizaban altas concentraciones tanto de IFN-α como de -ω, a pesar de las buenas respuestas de anticuerpos a la vacuna de ARN y de una capacidad normal para neutralizar el virus (Bastard et al., 2022, Sokal et al., 2023).
El desarrollo de una prueba de cribado sencilla en el entorno clínico para su implantación generalizada con un tiempo de respuesta rápido es justificable. Un resultado positivo en una prueba de este tipo en individuos sanos tendría implicaciones para la vacunación (gripe, COVID-19) y el seguimiento, y contraindicaría algunas otras vacunaciones (por ejemplo, la vacuna contra la fiebre amarilla YFV-17D). También tendría implicaciones para un tratamiento rápido y correcto en pacientes diagnosticados de infecciones víricas específicas. Por ejemplo, será interesante ver si el tratamiento con IFN-β es un enfoque factible (Bastard et al., 2020). Ensayos recientes con IFN-β han revelado pocas pruebas del beneficio de dicho tratamiento en pacientes hospitalizados (Kalil et al., 2021), aunque los ensayos en un entorno ambulatorio están justificados.
Un mecanismo general de enfermedad vírica
Recientemente se ha demostrado que los autoanticuerpos neutralizantes del IFN tipo I subyacen a la enfermedad grave por virus herpes simple o zóster en pacientes hospitalizados por COVID-19 (Busnadiego et al., 2022). Estos hallazgos concuerdan con el informe seminal de Ion Gresser y sus colegas sobre autoanticuerpos frente al IFN tipo I en una mujer de 77 años con herpes zóster diseminado (Pozzetto et al., 1984) y con la aparición de tales infecciones víricas en pacientes portadores de genotipos deletéreos de RAG1 o RAG2 o portadores de tales autoanticuerpos (Walter et al., 2015).
Recientemente se ha hecho una observación similar en una gran cohorte de pacientes con lupus eritematoso sistémico (Mathian et al., 2022). Además, un tercio de la pequeña serie de pacientes con reacciones adversas a la vacuna viva atenuada YFV-17D presentaba tales autoanticuerpos (Bastard et al., 2021). Sorprendentemente, entre estos pacientes había una mujer joven a la que posteriormente se diagnosticó lupus eritematoso sistémico, una anciana y un anciano. Estos tres grupos corren el riesgo de producir autoanticuerpos contra los IFN tipo I y ya se había demostrado que presentaban un mayor riesgo de reacciones adversas al YFV (Seligman et al., 2014). Por último, alrededor del 5% de los pacientes menores de 70 años eran portadores de tales autoanticuerpos y el riesgo estimado de gripe crítica aumentaba con la concentración y el número de IFN neutralizados (Zhang et al., 2022). Otras enfermedades víricas candidatas para las que los autoanticuerpos contra los IFN tipo I aumentan la susceptibilidad incluyen las infecciones víricas observadas en pacientes con errores innatos de la inmunidad a los IFN tipo I.
La evidencia de un papel de los autoanticuerpos contra los IFN tipo I ya está clara para al menos cuatro enfermedades víricas: neumonía crítica por COVID-19, neumonía gripal, reacciones adversas a la vacuna YFV-17D y herpes zóster recurrente o diseminado.
OBSERVACIONES FINALES
El descubrimiento de errores innatos de los IFN tipo I y de autoanticuerpos contra estas citocinas en al menos el 15% al 20% de los pacientes con neumonía crítica por COVID-19 sugirió un mecanismo general unificador de la enfermedad (Casanova y Abel, 2022; Zhang et al., 2022). El enigma de la COVID-19 «común» se descifró gracias a estudios previos realizados durante varias décadas en dos grupos de pacientes con fenotipos mendelianos «raros» y aparentemente opuestos: infecciosos y autoinmunes (Casanova y Abel, 2022). Este enfoque de raro a común, de paciente a población y de genético a mecanismo (Casanova y Abel, 2022, Zhang et al., 2022, Casanova y Abel, 2021) contrasta con otros planteamientos. El enfoque poblacional de la COVID-19, en el que esta «enfermedad común» se aborda mediante aproximaciones puramente matemáticas (estudios de asociación genética) o puramente inmunológicas (multiómica sanguínea o de mucosas), tuvo menos éxito. En lugar de detectar las causas inmunológicas de la enfermedad vírica, estos últimos estudios analizaron las respuestas inmunitarias al virus (Zhang et al., 2022). Y en lugar de detectar las causas genéticas de la enfermedad vírica en pacientes individuales, los primeros estudios detectaron modificadores comunes de la enfermedad a nivel poblacional.
Argumentamos que, con el enfoque de los “divisores”, centrado en pacientes individuales y «enfermedades raras», especialmente en pacientes jóvenes, con seres humanos individuales considerados como organismos individuales, puede ser posible, posteriormente, agrupar a pacientes con diferentes causas de enfermedad a través de mecanismos compartidos. Por el contrario, con el enfoque de los “agrupadores”, centrado en grandes poblaciones y «enfermedades comunes», sobre todo en poblaciones de edad avanzada, no es fácil dividir posteriormente a los pacientes en diferentes grupos, debido a la falta de causas y mecanismos de enfermedad identificados de forma inequívoca. Los estudios de valores atípicos «raros» constituyen un potente enfoque que puede utilizarse para guiar la exploración de enfermedades «comunes», ya sean víricas o de otro tipo (Casanova y Abel, 2022, Casanova y Abel, 2021; Lifton, 1996; Cohen et al., 2011; Goldstein y Browns, 2015).
AGRADECIMIENTOS
Agradecemos a Shen-Ying Zhang, Qian Zhang, Emmanuelle Jouanguy, Paul Bastard, Stéphanie Boisson-Dupuis, Vivien Béziat, Bertrand Boisson y Laurent Abel por la lectura crítica de una versión temprana del manuscrito. Agradecemos a Qian Zhang, Dana Liu, y Yelena Nemirovskaya por dibujar las figuras y editar las tablas.
MSA recibe apoyo de Chan-Zuckerberg Biohub, la Juvenile Diabetes Research Association, el Helmsley Charitable Trust, la Larry L. Hillblom Foundation, el National Institute of Diabetes and Digestive and Kidney Diseases y el National Institute of Allergy and Infectious Diseases (NIAID). JLC recibe apoyo del NIAID (R01AI088364, R01AI09983, R01AI127564, R01AI143810, R01AI163029, U19AI162568), el National Institute of Neurological Disorders and Stroke (R01NS072381), el National Center for Advancing Translational Sciences (UL1TR001866), el Howard Hughes Medical Institute, The Rockefeller University, la St. Giles Foundation, el Fisher Center for Alzheimer’s Research Foundation, la Meyer Foundation, la JPB Foundation, la French National Research Agency (ANR) bajo la subvención del Programa Futuro (ANR-10-IAHU-01), el Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), la French Foundation for Medical Research (FRM) (EQU201903007798), the HORIZON-HLTH-2021-DISEASE-04 Program (01057100; UNDINE), el Programa ANR-RHU COVIFERON (ANR-21-RHUS-08), la Square Foundation, Grandir–Fonds de solidarité pour l’enfance, la Fondation du Souffle, la SCOR Corporate Foundation for Science, el Ministerio Frances de Educación, Investigación e Innovación (MESRI-COVID-19), el Institut National de la Santé et de la Recherche Médicale (INSERM), REACTing-INSERM, y la Paris Cité University.
Conflictos de intereses
JLC es inventor de la patente PCT/US2021/042741 presentada el 22 de julio de 2021 por la Universidad Rockefeller que cubre el diagnóstico de la susceptibilidades y el tratamiento de enfermedad viral y vacuna viral, incluyendo COVID-19 y enfermedades asociadas a vacunas. JLC es miembro del comité asesor científico de ADMA Biologics Inc., Kymera Therapeutics y Elixiron Immunotherapeutics. MSA tiene acciones de Medtronic y Merck.