
INTRODUCCIÓN
La diabetes mellitus neonatal transitoria (DMNT) es una enfermedad rara que se caracteriza por la aparición de diabetes mellitus durante el primer año de vida, con una resolución espontánea de la misma en los primeros 18 meses. Sin embargo, la diabetes puede reaparecer durante la infancia, adolescencia o edad adulta. Su incidencia se estima en 1:300.000 (Wiedemann et al., 2010).
Existen tres formas de DMNT con una etiología genética diferente. La DMNT tipo 1 (DMNT1) o diabetes mellitus relacionada con el locus 6q24 [OMIM: 601410] es una enfermedad causada por alteraciones en los genes improntados localizados en el locus 6q24, mientras que la DMNT tipo 2 [OMIM: 610374] y tipo 3 [OMIM: 610582] resultan de alteraciones germinales en los genes ABCC8 y KCNJ11, respectivamente, que codifican para subunidades de un canal de potasio sensible a ATP y que se encuentran ambos en el locus 11p15.1 (Smith, 1996). También se han identificado alteraciones en los genes INS (Garin et al., 2010), HNF1B (Yorifuji et al., 2004; Edghill et al., 2006) y NEUROG3 (Rubio-Cabezas et al., 2014) asociadas a diabetes neonatal transitoria.
En este ensayo se describe la patogénesis de la DMNT1, una enfermedad producida por alteraciones en la impronta genómica. La impronta genómica es un proceso biológico que se caracteriza por el establecimiento de marcas epigenéticas, principalmente en regiones CpG, en las que se produce una metilación diferencial en ciertos loci durante la gametogénesis (e incluso a nivel postzigóstico), lo que resulta en una expresión génica específica del origen parental. Los loci improntados contienen genes con funciones clave en el desarrollo, crecimiento y metabolismo. Por ello, alteraciones en estas regiones improntadas pueden dar lugar a enfermedades de impronta (Monk et al., 2019).
DESCRIPCIÓN CLÍNICA
Los pacientes con DMNT1 desarrollan hiperglicemia durante su primera semana de vida, aunque la diabetes se resuelve de manera espontánea en los primeros 18 meses. Es importante recalcar que no se detectan autoanticuerpos frente a las células de los islotes pancreáticos en estos pacientes, lo que permite descartar un proceso autoinmune. Además, normalmente, no es necesaria la administración de insulina (Valerio et al., 2004; Boonen et al., 2013).
La presentación clínica de la diabetes va acompañada de deshidratación y ausencia de cetoacidosis. Otras manifestaciones clínicas frecuentes son un retraso de crecimiento intrauterino durante el embarazo, y un peso (pero no talla) al nacer inferior a la media, debido a los niveles insuficientes de insulina en el útero. Con el tiempo, una vez la diabetes ha remitido, los parámetros de crecimiento vuelven a la normalidad. Se han reportado también algunas anomalías congénitas en infantes con DMNT1, como macroglosia y hernia umbilical (Temple y Mackay, 2005; Shield, 2000; Temple et al., 2000; Rica et al., 2007; Yorifuji et al., 2018). Asimismo, los pacientes con alteraciones de impronta multi-loci pueden presentar retraso en el desarrollo, afectación neurológica, sordera, hipotonía, enfermedades cardiacas congénitas y malformaciones renales (Temple y Mackay, 2005).
Frecuentemente, los episodios de hiperglicemia vuelven a aparecer durante la infancia o la adolescencia, especialmente durante el transcurso de una enfermedad (Shield et al., 1997). Además, se han reportado casos donde los pacientes desarrollan hipoglicemia hiperinsulínica tras la remisión de la diabetes, que podría deberse a un exceso en la secreción de insulina (Flanagan et al., 2013). Desafortunadamente, una considerable proporción de pacientes con DMNT1 adquiere una diabetes permanente durante la adolescencia o edad adulta joven, que podría ser consecuencia de una disfunción pancreática residual. Esta diabetes se caracteriza por una disminución de la secreción de insulina, ausencia de anticuerpos de islotes pancreáticos y un peso corporal normal, con similitud a la diabetes MODY (diabetes de la edad madura que se presenta en el joven) (Temple y Mackay, 2005; Shield, 2000; Temple et al., 2000; Yorifuji et al., 2018). En dicho caso, la diabetes se puede tratar con insulina, sulfonilureas o controlando la dieta. Ante una falta de respuesta al tratamiento, otras alternativas para disminuir los niveles de glucosa en sangre son el uso de inhibidores de la β-glucosidasa, gliptinas o glinidas (Yorifuji et al., 2018).
ETIOLOGÍA CLÍNICA
La primera persona que sugirió que la DMNT1 podría ser una enfermedad de impronta fue David Haig en 1994 (Haig, 1994), quien además postuló anteriormente que los genes de la insulina estaban improntados. En 1995, Temple y sus colaboradores corroboraban esta hipótesis al descubrir que los pacientes con DMNT1 presentaban con frecuencia una disomía uniparental paterna del cromosoma 6. Este descubrimiento sugería la presencia de un gen improntado que estaría implicado en la función de las células beta pancreáticas durante el desarrollo fetal y neonatal (Temple et al., 1995).
Un año después, el grupo de investigación de Temple describía otros casos de DMNT1 debidos a duplicaciones del cromosoma 6, cuyo patrón de herencia apoyaba la hipótesis de una enfermedad de impronta, ya que la enfermedad solo se manifestaba en pacientes con duplicaciones paternas mientras que, si portaban duplicaciones maternas, eran asintomáticos. Sus experimentos sugerían que el gen causante de la DMNT1 se encontraba en la región cromosómica 6q22.33—q23.3 y que presentaba impronta materna. De esta manera, si solo se expresa la copia paterna del gen improntado, la disomía uniparental paterna y la duplicación paterna del cromosoma 6 resultarían ambos en la sobreexpresión de dicho gen y, por tanto, en el mismo fenotipo (Temple et al., 1996).
En el año 2000, el mismo grupo refinaba la región improntada a un área de 300-400 kilobases situada en la región cromosómica 6q24 que incluía seis islas CpG. De hecho, descubrieron que, en una de estas islas, los pacientes con disomía uniparental paterna y los controles presentaban un patrón de metilación diferente: en los pacientes se encontraba hipometilada, con dos copias paternas no metiladas, mientras que los controles tenían un alelo materno metilado y un alelo paterno no metilado. Además, estos investigadores describieron un tercer mecanismo molecular implicado en el desarrollo de la DMNT1, que consistía en la pérdida de la metilación en el alelo materno, lo que también conducía a una sobreexpresión del gen improntado. Estos descubrimientos confirmaban la presencia de un locus improntado en la región 6q24 involucrado en la DMNT1 (Gardner et al., 2000).
El locus improntado 6q24 contiene un promotor compartido por dos genes, PLAGL1 (pleiomorphic adenoma gene-like 1), también conocido como ZAC1 (zinc finger protein associated with apoptosis and cell-cycle arrest), e HYMAI (hydatidiform mole associated and imprinted). En el año 2000, se descubría que ambos genes estaban improntados en humanos, ya que existía una isla CpG adyacente a dichos genes que estaba diferencialmente metilada y únicamente se expresaba la copia paterna de ambos genes (Arima et al., 2000; Kamiya et al., 2000). Respaldando esta hipótesis, se observó que, en un paciente con DMNT1 con pérdida de metilación en el alelo materno, la expresión de PLAGL1 e HYMAI ya no era exclusivamente paterna, sino que ambos alelos se expresaban (Mackay et al., 2002).
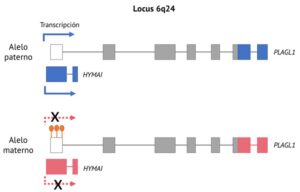
En resumen, la DMNT1 está causada por alteraciones en el locus improntado 6q24 que conducen a la sobreexpresión de los genes improntados PLAGL1 e HYMAI. Este locus incluye una región diferencialmente metilada, estando metilada en el alelo materno y no metilada en el paterno, lo que resulta en una expresión específica del alelo paterno de los genes PLAGL1 e HYMAI (Figura 1). Actualmente, se conocen tres causas genéticas para la DMNT1: la disomía uniparental paterna del cromosoma 6 (41% de los casos), que puede afectar a un segmento o al cromosoma entero; la duplicación paterna de la región 6q24 (33%), que puede ser de novo, heredada o causada por alteraciones cromosómicas, y la pérdida de metilación materna de la región diferencialmente metilada del locus 6q24 (~26%). Este último mecanismo molecular se conoce como epimutación, que puede ser primaria, debido a errores reproductivos o factores ambientales, o secundaria, causada por alteraciones genéticas en elementos cis o factores trans (Mackay y Temple, 2010; Mackay et al., 2014). Sin embargo, todavía no se conoce si uno o los dos genes PLAGL1 e HYMAI son los genes causantes de la DMNT1.
Cabe mencionar que la manifestación clínica de la diabetes no suele variar en función de la etiología genética del paciente. Sin embargo, algunos pacientes con DMNT1 no desarrollan diabetes neonatal, lo que implica una penetrancia incompleta (Valerio et al., 2004; Bliek et al., 2009; Boonen et al., 2013). Otras características fenotípicas sí pueden variar en función del mecanismo molecular. No obstante, pacientes que presentan la misma causa genética pueden desarrollar diferentes alteraciones fenotípicas, por lo que la correlación entre el genotipo y fenotipo no es inmediata (Marquis et al., 2000). Por ejemplo, es más probable que individuos con disomía uniparental paterna desarrollen problemas relacionados con enfermedades recesivas del cromosoma 6. Por otro lado, los pacientes con duplicaciones normalmente no presentan otras complicaciones. Sin embargo, duplicaciones que afecten a varios loci pueden producir otras alteraciones, como retraso mental o disformismo. En cuanto a los pacientes con epimutaciones, aquellos en los que únicamente existe una hipometilación del locus de DMNT1 tienen una presentación clínica similar a otros casos de DMNT1, mientras que los individuos con alteraciones de impronta multi-loci presentan un mayor número de complicaciones (Mackay y Temple, 2010; Yorifuji et al., 2018).
DNMT1 CON ALTERACIÓN DE LA IMPRONTA MULTI-LOCI
Algunos pacientes con DMNT1 presentan alteraciones en otras regiones genómicas improntadas además del locus 6q24. En este contexto, en el año 2005, Arima y su grupo observaron que algunos individuos con DMNT1 presentaban una hipometilación de la región improntada KCNQ1OT1:TSS-DMR en 11p15, asociada con el síndrome de Beckwith-Wiedemann (Arima et al., 2005). Otro estudio posterior corroboraba la existencia de una hipometilación materna en mosaico en múltiples loci improntados (GRB10:alt-TSS-DMR en 7p21, MEST:alt-TSS-DMR en 7q32, KCNQ1OT1:TSS-DMR en 11p15 y PEG3:TSS-DMR en 19q13) en pacientes con DMNT1, que resultaban en manifestaciones clínicas mucho más diversas. Estos descubrimientos sugerían que este síndrome podría estar causado por un fallo en el mantenimiento de la metilación durante una etapa muy temprana del zigoto (Mackay et al., 2006).
Tras estas observaciones, se llevó a cabo un estudio que evaluaba una cohorte de 13 pacientes con hipometilación materna de múltiples loci improntados. Dicho estudio reveló la existencia de variantes patogénicas en homocigosis en el gen ZFP57 en seis familias consanguíneas y un caso de heterocigosis compuesta. Aparentemente, todos los individuos con hipometilación en el locus 6q24 también presentaban una hipometilación materna en los loci PEG3:TSS-DMR y GRB10:alt-TSS-DMR, mientras que no se detectaron alteraciones en ZFP57 en aquellos individuos que únicamente presentaban pérdida de la metilación en el locus 6q24 (Mackay et al., 2008). Posteriormente, la investigación clínica y molecular de 10 familias con DMNT1 y variantes patogénicas en ZFP57 corroboraba dichas observaciones. Estos estudios revelaban una pérdida de metilación completa en el locus 6q24, mientras que el grado de hipometilación era variable en los otros loci con impronta materna (Boonen et al., 2013).
El gen ZFP57 codifica para una proteína de dedos de zinc que se expresa muy pronto durante la embriogénesis. Las variantes patogénicas identificadas eran o bien truncantes o bien con cambio de sentido afectando a dominios funcionales importantes. Debido a la asociación de estas alteraciones con la hipometilación materna en mosaico de regiones improntadas, se cree que ZFP57 tiene un papel en el mantenimiento de la metilación del ADN durante el desarrollo temprano (Mackay et al., 2008).
No obstante, solo un 50% de los casos de DMNT1 con alteraciones de la impronta multi-loci están causados por variantes recesivas bialélicas en ZFP57, mientras que la causa del 50% restante se desconoce. Alteraciones en otros factores trans involucrados en el establecimiento, mantenimiento y/o eliminación de marcas epigenéticas, o alteraciones de elementos cis podrían también resultar en epimutaciones secundarias. Por otra parte, también podrían ocurrir epimutaciones primarias (no relacionadas con alteraciones genéticas) o bien al azar o causadas por factores ambientales. Como ejemplo de ello, un tema de debate actual es la posibilidad de que las técnicas de reproducción asistida causen alteraciones epigenéticas o bien en el gameto o en el embrión, debido a la elevada frecuencia de enfermedades de impronta en niños nacidos por medio de estas técnicas (Pinborg et al., 2016; Uk et al., 2018). Otros factores ambientales que podrían causar alteraciones epigenéticas son deficiencias nutricionales, alteraciones metabólicas o exposición a disruptores endocrinos durante el embarazo (Monk et al., 2019).
CAUSAS DE LA DIABETES
El estudio de la funcionalidad de las células beta pancreáticas en modelos de enfermedad y pacientes ha sido clave para conseguir un mejor entendimiento de cómo un fallo en la impronta genómica conduce a una disfunción pancreática y a hiperglicemia. Ma y colaboradores desarrollaron un modelo de ratón transgénico de DMNT1 con sobreexpresión del locus 6q24, a partir de múltiples copias de un clon BAC (cromosoma artificial bacteriano) conteniendo los genes PLAGL1 e HYMAI, con el fin de evaluar los efectos de la DMNT1 en la función pancreática. Este modelo reproducía los fenotipos característicos de la DMNT1 ya que, únicamente cuando el transgén se heredaba del padre, las crías de ratón presentaban hiperglicemia. Esta hiperglicemia desaparecía en el ratón joven y volvía a aparecer más tarde en la etapa adulta. Observaron que la hiperglicemia neonatal en los ratones era resultado de una síntesis y secreción deficiente de insulina, aunque la masa celular de células beta pancreáticas era normal. Sin embargo, en los ratones jóvenes con un contenido de insulina normal, detectaron un aumento en el número de células beta pancreáticas. Además, el estudio de dicho modelo de ratón de DMNT1 a nivel embrionario revelaba una alteración en el desarrollo del páncreas, consistente con la disminución de la expresión de factores de transcripción claves en el desarrollo pancreático (Ma et al., 2004). Estas observaciones demostraban que el locus 6q24 está implicado en el desarrollo del páncreas y en la función de las células beta pancreáticas.
Por otro lado, un estudio post mortem de un neonato con DMNT1 que falleció a los 16 días reveló una ausencia total de células beta productoras de insulina, lo que sugería una implicación del gen causante de la DMNT1 en el desarrollo de estas células (Blum et al., 1993). Otros investigadores exploraron la funcionalidad de las células beta en pacientes con recaída y observaron que dichas células no respondían a la estimulación con glucosa, pero sí secretaban insulina tras la estimulación con glucagón. La producción y secreción de insulina tras el estímulo de glucosa o glucagón está mediada por dos vías distintas, lo que indicaría que solo la vía de secreción de insulina en respuesta a glucosa está afectada en estos pacientes (Valerio et al., 2004).
Como se ha mencionado anteriormente, la alteración de la impronta en el locus 6q24 de la DMNT1 da lugar a la sobreexpresión de los genes improntados PLAGL1 e HYMAI. El gen PLAGL1 codifica para un factor de transcripción de dedos de zinc que participa en el arresto del ciclo celular y apoptosis actuando como un coactivador de p53. Se ha especulado que la sobreexpresión de PLAGL1 durante el desarrollo del páncreas podría causar la apoptosis de las células beta pancreáticas (Mackay et al., 2002). Por otro lado, HYMAI es un transcrito de ARN no codificante, expresado de manera ubicua, con una función desconocida.
Ambos genes PLAGL1 e HYMAI comparten el mismo promotor improntado. Sin embargo, PLAGL1 presenta otro promotor no improntado. La expresión génica bialélica de PLAGL1 a partir de este último promotor se ha detectado en el páncreas y en otros tejidos (Valleley, Cordery y Bonthron, 2007). Por tanto, se ha sugerido que la DMNT1 podría deberse a la sobreexpresión de PLAGL1 en el feto en un tejido y en una etapa del desarrollo embrionario específicos, que puede después remitir debido al cambio al promotor no improntado. En dicho contexto, la reaparición de la diabetes podría explicarse por una sobreexpresión residual crónica del gen PLAGL1 a partir del promotor improntado o por una masa reducida de células beta pancreáticas. Como consecuencia, PLAGL1 es el principal candidato como gen causante de la DMNT1, aunque no se puede excluir la implicación del gen HYMAI, ya que se han descubierto otros transcritos no codificantes con un papel importante en enfermedades de impronta (Mackay y Temple, 2010).
En consecuencia, se han llevado a cabo varios estudios para tratar de determinar el rol del gen PLAGL1 en la presentación de la diabetes. De acuerdo con descubrimientos previos, estudios llevados a cabo en una línea celular de células beta pancreáticas de ratón revelaban que la sobreexpresión de PLAGL1 afecta negativamente a la producción y secreción de insulina tras la estimulación con glucosa (Du et al., 2012). El primer gen diana de PLAGL1 que se identificó fue el receptor del polipéptido activador de la adenilato ciclasa de la pituitaria (PACAP). PLAGL1 reduce la expresión de dicho receptor, cuya función es promover la secreción de insulina estimulada por glucosa por los islotes pancreáticos (Hoffman et al., 1998). Además, la sobreexpresión de PLAGL1 en células beta murinas permitió la identificación de otro gen diana, RASGRF1, cuya expresión está también regulada negativamente por PLAGL1, conduciendo a una disminución de la secreción de insulina (Hoffmann y Spengler, 2012). Otros ensayos de expresión génica posteriores han revelado un mayor número de genes diana de PLAGL1 involucrados en la funcionalidad de las células beta. Hoy en día se conoce que la sobreexpresión de PLAGL1 conduce a una disminución de la secreción de insulina y a una reducida proliferación de las células beta por medio del aumento de la expresión de PPARγ, TCF4 y SOCS3, y la disminución de la expresión del receptor de PACAP y RASGRF1 (Hoffmann y Spengler, 2015).
Estos descubrimientos parecen explicar cómo la sobreexpresión de PLAGL1 produce una diabetes neonatal. No obstante, se requieren más estudios para esclarecer la causa de la remisión espontánea de la diabetes así como los mecanismos que desencadenan la recaída posterior durante la adolescencia o edad adulta.
MECANISMOS MOLECULARES DE LA ALTERACIÓN DE LA IMPRONTA
El conocimiento de los mecanismos moleculares involucrados en las enfermedades de impronta ha avanzado considerablemente gracias al estudio de modelos animales. En el contexto de la DMNT1, Arima et al. descubrieron que la región 6q24 era sintética a la región cromosómica 10A en ratón, donde un análisis de expresión en embriones de ratón revelaba una expresión exclusivamente paterna del gen Plagl1 en todos los tejidos analizados, sugiriendo que este gen también estaba improntado en ratones (Arima et al., 2000). Asimismo, el mismo grupo descubría posteriormente una posible región reguladora de esta región improntada en el locus sintético de ratón (Arima et al., 2001).
Además, estudios de la línea germinal de ratón revelaban que la metilación del ADN de la región improntada se adquiría en la línea germinal femenina durante un estadio temprano, coincidiendo con la pérdida de expresión de Plagl1 en los ovocitos. En consonancia, estudios en ovocitos humanos confirmaban también que la impronta en 6q24 es un evento temprano, ya que la metilación en el ADN se puede detectar en ovocitos de folículos primordiales y primarios (Arima y Wake, 2006).
Por otro lado, Li y colaboradores emplearon un modelo de ratón con el fin de elucidar la función del factor Zfp57 en el establecimiento y mantenimiento de la metilación en regiones improntadas. Mientras que la pérdida de la función de Zfp57 en el zigoto resultaba en una pérdida parcial de la metilación diferencial del ADN y letalidad parcial, la pérdida de las funciones de Zfp57 en el gameto femenino y en el zigoto daba lugar a una pérdida total de la metilación diferencial, y por consiguiente, a una mortalidad embrionaria elevada. El mismo estudió revelaba, además, que Zfp57 es importante para el establecimiento de la impronta en ciertos loci en la línea germinal femenina y para el mantenimiento de la metilación del ADN en loci improntados paternos (MEG3/DLK1:IG-DMR) y maternos (MEST:alt-TSS-DMR, PEG3:TSS-DMR y NNAT:TSS-DMR) durante el desarrollo embrionario (Li et al., 2008). Estas observaciones corroboraban el hecho de que variantes patogénicas en ZFP57 resulten en hipometilación de varios loci improntados.
Asimismo, Quenneville y colaboradores estudiaron el rol de Zfp57 y su cofactor Kap1 en células troncales embrionarias de ratón, y observaron que ambos se unen a alelos metilados de regiones del control de la impronta y mantienen la metilación del ADN, las modificaciones de histonas y la heterocromatina (Quenneville et al., 2011). Por tanto, estas observaciones indicarían que Zfp57 protege a los loci improntados de la inestabilidad epigenética que existe en la embriogénesis temprana.
Curiosamente, los estudios en modelo de ratón han revelado que Zfp57 es importante para mantener la impronta materna y paterna en varios genes, pero solo la hipometilación materna se ha observado en humanos. Esto podría explicarse por el hecho de que la cohorte de pacientes evaluada únicamente incluye pacientes con DMNT1. Sin embargo, se requieren más estudios enfocados a caracterizar la función de ZFP57 en modelos humanos (Mackay y Temple, 2010).
DIAGNÓSTICO Y RIESGO DE RECURRENCIA
Todos los pacientes con sospecha de DMNT1 deberían someterse a un consejo genético y test genético para determinar el mecanismo molecular responsable de la enfermedad (Tabla 1). Aunque la manifestación de la diabetes es independiente del mecanismo molecular, es importante que se defina la etiología genética de la enfermedad, ya que ésta es clave para determinar el riesgo de recurrencia y proporcionar al paciente y sus familiares un adecuado consejo genético.
Tests genéticos moleculares para determinar la etiología genética de pacientes con DMNT1.
| Alteración | Tecnología | Objetivo | Casos |
| Hipometilación en el locus 6q24 | Secuenciación por bisulfito
Southern Blot específico de metilación MS-MLPA PCR específica de metilación |
Establecer diagnóstico | 100% |
| UPD | Análisis de marcadores microsatélites polimórficos
Microarrays basados en SNP |
Determinar mecanismo molecular | ~41% |
| Duplicaciones | PCR cuantitativa
PCR de largo alcance MS-MLPA Microarrays basados en oligonucleótidos Microarrays basados en SNP |
Determinar mecanismo molecular | ~29% |
| Alteraciones del gen ZFP57 | Secuenciación
PCR cuantitativa PCR de largo alcance MLPA |
Determinar mecanismo molecular | ~9% |
Adaptado de Temple y Mackay (2005).
MS-MLPA, amplificación múltiple, específica de metilación, de sondas ligadas. PCR, reacción en cadena de la polimerasa. SNP, polimorfismo de nucleótido único. UPD, disomía uniparental.
Las diferentes alteraciones en la región 6q24 tienen como consecuencia la hipometilación de este locus. Dicha hipometilación se puede detectar por medio de estudios de metilación del ADN mediante la técnica de secuenciación por bisulfito o la técnica de MS-MLPA (amplificación múltiple, específica de metilación, de sondas ligadas), entre otros métodos. Estas técnicas permiten confirmar el diagnóstico de la DMNT1. Sin embargo, puede ser necesario emplear otras pruebas que permiten discernir la etiología genética, y por tanto, determinar el correspondiente riesgo de recurrencia.
Las duplicaciones del locus 6q24 pueden analizarse mediante microarrays cromosómicos, bien microarrays de oligonucleótidos o microarrays de SNP (polimorfismo de nucleótido único), o con métodos alternativos, como el uso de la PCR cuantitativa o mediante la técnica de MS-MLPA, entre otros. Para los estudios de disomía uniparental paterna, ya sea parcial o completa, puede emplearse el análisis de segregación de marcadores microsatélites polimórficos distribuidos a lo largo del cromosoma implicado o los microarrays de SNP (Temple y Mackay, 2005).
En el caso de que no se identifiquen ni duplicaciones ni disomía, se concluye que el mecanismo molecular causante de la DMNT1 es la presencia de epimutaciones. Si estas epimutaciones afectan a otros loci, es importante intentar discernir si tienen una causa genética, por lo que se procede a realizar un test genético para analizar la secuencia del gen ZFP57 así como posibles duplicaciones o deleciones, con el fin de detectar posibles variantes patogénicas en dicho gen (Temple y Mackay, 2005).
Por otro lado, la recurrencia de la DMNT1 en futuras generaciones depende del mecanismo molecular. Los descendientes de individuos con disomía uniparental paterna no presentan riesgo de recurrencia, ya que la disomía uniparental suele deberse a un error al azar durante la meiosis, a menos que los progenitores presenten otras alteraciones cromosómicas (Mackay y Temple, 2010).
En aquellos pacientes con duplicaciones, es crucial conocer si la duplicación es heredada o de novo. Si es heredada del padre, la duplicación se transmitirá de manera dominante. De esta manera, los descendientes de padres portadores tendrían 50% de riesgo de heredar la enfermedad, mientras que las madres portadoras no transmiten la enfermedad (Mackay y Temple, 2010).
Respecto a los pacientes que presentan variantes patogénicas bialélicas del factor ZFP57, todos los descendientes serán, como mínimo, portadores. Si la pareja del individuo afectado es asimismo portadora de una alteración en ZFP57, cada uno de sus descendientes tiene un 50% de probabilidades de heredar ambos alelos alterados y, por tanto, desarrollarían alteraciones de la impronta multi-loci, y un 50% de probabilidades de ser portador asíntomático.
Finalmente, en aquellos pacientes con pérdida de metilación únicamente en el locus de DMNT1 o con hipometilación materna multi-loci sin alteraciones en ZFP57, el riesgo de recurrencia no se conoce (Temple y Mackay, 2005; Mackay y Temple, 2010).
CONCLUSIONES
La DMNT1 es una enfermedad de impronta cuyos mecanismos genéticos y epigenéticos se han descrito por medio de la investigación genética de pacientes y familiares. Asimismo, modelos de ratón y estudios humanos han sido cruciales a la hora de comprender cómo este fallo en la impronta genómica conduce a una disfunción de las células beta pancreáticas y para entender los mecanismos moleculares de la impronta. Sin embargo, se requiere una mayor investigación para clarificar porqué la mayoría de los pacientes recaen y para descifrar las causas de las epimutaciones. Todo este conocimiento contribuirá a un mejor manejo clínico de los pacientes y a expandir nuestro conocimiento de las enfermedades de impronta. En resumen, la DMNT1 es un gran ejemplo de la interacción entre genética, epigenética y ambiente en la diabetes mellitus.


