
EL DISTROGLICANO
El distroglicano (DG) fue descrito por primera vez en células neurales murinas híbridas neuroblastoma ⨯ glioma de la línea NG108-15, en fibroblastos embrionarios de ratón de la línea 3T3 y en cerebro embrionario de pollo como la glicoproteína predominante de la membrana plasmática capaz de unirse a la laminina (Smalheiser y Schwartz, 1987), el componente principal de la lámina basal de la matriz extracelular (ECM) (Durbeej, 2010). El DG está codificado por el gen DAG1 (OMIM 128239), localizado en el cromosoma 3p21 y constituido por dos exones separados por un largo intrón. Este gen se transcribe en un único mRNA de 5748 bases cuyo producto proteico contiene 895 aminoácidos (aas) y presenta una masa molecular de 97,5 kDa en nuestra especie. Este polipéptido sufre una escisión postraduccional de forma autocatalítica en el residuo Ser-654 para producir las subunidades denominadas α-distroglicano (α-DG) y β-distroglicano (β-DG), las cuales permanecen asociadas de forma no covalente en la membrana plasmática (Ibraghimov-Beskrovnaya et al., 1992; Holt et al., 2000; Akhavan et al., 2008; Oppizzi et al., 2008). La secuencia de aminoácidos adyacente al sitio de escisión no está conservada entre vertebrados e invertebrados. Sin embargo, se ha demostrado que la sobreexpresión de un propéptido alterado en este sitio, portador de la sustitución S654A (Ala en lugar de Ser-654), impide su procesamiento postraduccional y provoca cambios distróficos en el músculo esquelético de ratones transgénicos (Jayasinha et al., 2003). Ello sugiere que la escisión del DG en sus dos subunidades es crucial para la función y el desarrollo normal de (al menos) los músculos esqueléticos.
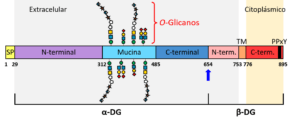
La estructura primaria del DG, a diferencia del sitio de escisión, está altamente conservada en vertebrados, sugiriendo que su función y estructura se han mantenido durante la evolución de los cordados (Parsons et al., 2002). Esta glicoproteína se expresa en una amplia variedad de tejidos fetales y adultos, entre los que se encuentran el músculo, los nervios, el tejido adiposo, el epitelio, el endotelio y la sangre, siendo prevalente en el músculo esquelético y el cerebro (Ibraghimov-Beskrovnaya et al., 1992, 1993; Durbeej et al., 1995, 1998; Durbeej y Campbell, 1999). Los primeros 29 aminoácidos traducidos del propéptido son predominantemente hidrofóbicos y constituyen el péptido señal del DG (Barresi y Campbell, 2006) (Fig. 1), cuya función general es determinar la vía de transporte, la localización y la eficiencia de secreción de la proteína. Una vez eliminado el péptido señal, la subunidad α-DG madura (aas 30-653; ~74 kDa) constituye un polipéptido extracelular asociado a la membrana celular, formado por dos dominios globulares N- y C-terminales separados por una región de tipo mucina rica en residuos de prolina, serina y treonina altamente O-glicosilados, y que además contiene tres sitios de N-glicosilación (Ervasti y Campbell, 1991; Ibraghimov-Beskrovnaya et al., 1992; Brancaccio et al., 1995, 1997) (Fig. 1). En cambio, la subunidad β-DG (aas 654-895; ~43 kDa) contiene un único residuo N-glicosilado y carece de sitios de O-glicosilación (Ibraghimov-Beskrovnaya et al., 1992; Oppizzi et al., 2008). Este polipéptido está constituido por un dominio N-terminal que contiene una región transmembrana, y un dominio C-terminal citoplásmico enriquecido en residuos de prolina y que alberga un motivo PPxY de unión a la distrofina (Brancaccio et al., 1995; Jung et al., 1995) (Fig. 1).
El DG forma parte del llamado complejo distrofina-glicoproteína (DGC), el cual es responsable de unir determinadas proteínas de la ECM a la actina del citoesqueleto a través de la distrofina en células del músculo esquelético y tejidos no musculares (Winder, 2001; Ervasti y Sonnemann 2008; Dobson et al., 2013). Este complejo está también compuesto por el sarcoglicano (en el músculo), distrobrevinas, sintrofinas y sarcospán, e incluye otros elementos periféricos o proteínas asociadas, como son la óxido nítrico sintasa neuronal (nNOS o NOS1) y la caveolina 3 (Cav-3) (Ervasti y Campbell, 1991, 1993; Durbeej y Campbell, 1999) (Fig. 2).
El DGC contribuye a la estabilidad estructural de la membrana plasmática de la célula muscular (sarcolema) durante los ciclos de contracción y relajación, protegiendo al músculo del daño en la membrana inducido por estrés (Petrof et al., 1993; Campbell, 1995; Cohn, 2005). Así, alteraciones en el DG o el sarcoglicano provocan inestabilidad y contracción en el sarcolema, originando un incremento en el flujo entrante de Ca2+ en este último (Alderton y Steinhardt, 2000) que puede causar la necrosis de las fibras musculares, característica principal de las distrofias musculares (Bushby, 2000; Cohn y Campbell, 2000). Además, el DGC está implicado en otras funciones, como son el desarrollo temprano del embrión de ratón (Williamson et al., 1997), la estructura y función del sistema nervioso central (CNS) (Moore et al., 2002), la mielinización y establecimiento de la estructura nodal de los nervios periféricos (Cohn et al., 2002; Saito et al., 2003), la morfogénesis epitelial (Durbeej y Ekblom, 1997; Durbeej et al., 2001), la adhesión celular (Matsumura et al., 1997; Bello et al., 2015), la sinaptogénesis (Jacobson et al., 1998; Montanaro et al., 1998), la señalización intracelular en el músculo esquelético (Langenbach y Rando, 2002; Spence et al., 2004) y el mantenimiento de la organización estructural de la retina (Clements et al., 2017).
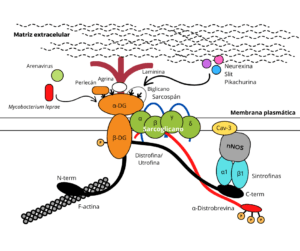
En el DGC el α-DG es responsable de la unión de la célula a la ECM, al interactuar de forma no covalente por un lado con el dominio N-terminal del β-DG, y de forma dependiente de Ca2+ por otro con proteínas componentes de la ECM, como son la laminina, agrina, perlecán y biglicano en tejidos musculares y nerviosos, neurexina y slit en el cerebro, y pikachurina en la retina, a través de los O-manosilglicanos unidos a su dominio de tipo mucina (Huang et al., 2000; Barresi y Campbell, 2006; Samwald, 2007; Sato et al., 2008; Nickolls y Bönnemann, 2018) (Fig. 2). Es esencial que el α-DG esté altamente glicosilado para su correcto funcionamiento como receptor en la superficie de las fibras musculares y de las células del sistema nervioso (Barresi y Campbell, 2006), pero también para llevar a cabo otras funciones en las que se ha demostrado más recientemente que está involucrado, como son la embriogénesis, procesos neoplásicos y progresión tumoral, transducción de señales, y adhesión y entrada de patógenos a sus células hospedadoras. En este último caso, el α-DG glicosilado interacciona extracelularmente, actuando como receptor, con determinados agentes infecciosos, como son algunos miembros de la familia arenavirus, entre ellos el responsable de la coriomeningitis linfocítica (LCMV) y el de la fiebre de Lassa (LFV), y la bacteria Mycobacterium leprae en las células de Schwann (Winder, 2001; Bozzi et al., 2009; Yoshida-Moriguchi y Campbell, 2015).
Por su parte, en el DGC la porción citosólica del β-DG, es decir, su dominio C-terminal (Fig. 1), está anclada al citoesqueleto de actina mediante su interacción, bien con la distrofina en el músculo, bien con su parálogo utrofina en tejidos no musculares y en la unión neuromuscular (Bozzi et al., 2009; Montanaro y Martin, 2011) (Fig. 2). Este dominio contiene el llamado motivo PPxY892, el cual se une al dominio WW de la distrofina o de la utrofina (Fig. 2), y al dominio SH3 de la llamada proteína 2 unida a receptores de factores de crecimiento (Grb2). El residuo Tyr-892 presente en dicho motivo puede ser fosforilado, provocando que el β-DG se disocie de los dominios WW y SH3, perdiendo así su nexo con el citoesqueleto y ganando afinidad por proteínas adaptadoras que contienen el dominio SH2, como son Grb2 y c-Src. Ello le permite al β-DG desempeñar una función clave en la modulación de diversas rutas de señalización intracelular (Bello et al., 2015).
LA GLICOSILACIÓN DEL α-DISTROGLICANO
El DG es una proteína altamente glicosilada en el dominio de tipo mucina de su subunidad α, el cual contiene un elevado número de sitios de O-glicosilación y tres sitios de N-glicosilación, mientras que la subunidad β solo presenta un único residuo N-glicosilado (Ervasti y Campbell, 1991; Ibraghimov-Beskrovnaya et al., 1992; Brancaccio et al., 1995, 1997; Oppizzi et al., 2008). Como resultado de la extensa glicosilación de su polipéptido central, el α-DG, del cual se predice una masa molecular de aproximadamente 74 kDa, se detecta mediante Western blotting como una banda que varía ampliamente de tamaño entre 100 y 190 kDa dependiendo del tejido estudiado (Ibraghimov-Beskrovnaya et al., 1992; Matsumura et al. 1997; Cohn, 2005; Barresi y Campbell, 2006). La eliminación de los N-glicanos mediante el tratamiento del α-DG con N-glicanasas disminuye su masa molecular en tan solo 4 kDa (Ervasti y Campbell, 1991), por lo que la amplia variabilidad de tamaños citada es probablemente el resultado de diferencias en el grado de O-glicosilación de su dominio de tipo mucina que se originan durante el desarrollo (Barresi y Campbell, 2006). Adicionalmente, otros estudios revelan que los N-glicanos no tienen relevancia para la función del α-DG como receptor de la ECM, lo que indica que dichos azúcares no son necesarios para la unión a sus ligandos. En cambio, la completa desglicosilación del α-DG tiene como resultado una pérdida total de su actividad de unión a ligandos (Ervasti y Campbell, 1993). Por ello, los O-glicanos unidos al dominio de tipo mucina son los responsables de las interacciones del α-DG con otras proteínas de la ECM (Barresi y Campbell, 2006).
En el dominio de tipo mucina del α-DG existen alrededor de 50 residuos de serina y treonina susceptibles de ser O-glicosilados. La estructura básica de las cadenas de O-glicanos del α-DG es una mezcla de polisacáridos que comienzan por N-acetilgalactosamina (GalNAc) o manosa (Man), siendo estos últimos los más abundantes (Martin, 2003). La O-manosilglicosilación de proteínas es un proceso altamente conservado evolutivamente desde hongos hasta mamíferos (Panin y Wells, 2014), siendo el α-DG la primera proteína confirmada en sufrir esta particular modificación postraduccional (Chiba et al., 1997; Wells, 2013; Sheikh et al., 2017). Los O-manosilglicanos unidos al α-DG son cadenas heterogéneas, catalizadas por distintas glicosiltransferasas que actúan secuencialmente (Martin, 2007), que se clasifican según el enlace establecido entre la N-acetilglucosamina (GlcNAc) y la O-manosa, esta unida a una serina o treonina del α-DG, en tres núcleos estructurales denominados M1, M2 y M3 (Yoshida-Moriguchi et al., 2013; Endo, 2015, 2019; Sheikh et al., 2017).
La O-manosilglicosilación del α-DG, así como la síntesis de los tres núcleos estructurales descritos, comienza en la cara citosólica del retículo endoplásmico con la generación de una molécula de dolicol-fosfato-manosa (Dol-P-Man), donadora de manosa, por la actividad del complejo dolicol-fosfato manosiltransferasa, constituido por tres subunidades denominadas DPM1, 2 y 3 (Maeda et al., 2000; Lefeber et al., 2009; Yang et al., 2013) (Fig. 3). A continuación, las enzimas conocidas como proteína O-manosiltransferasas 1 (POMT1) y 2 (POMT2), las cuales forman un heterocomplejo, son las responsables de transferir una unidad de manosa desde la Dol-P-Man al grupo OH de los residuos de serina o treonina del dominio de tipo mucina del α-DG (Manya et al., 2004) (Fig. 3).
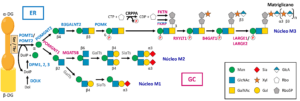
El primer paso en la elongación de la O-manosa del núcleo M1 es la adición de una molécula de GlcNAc mediante un enlace β1,2, catalizada en el complejo de Golgi por la enzima denominada proteína O-manosa β1,2-N-acetilglucosaminiltransferasa 1 (POMGNT1), la cual utiliza como sustrato UDP-GlcNAc para originar el disacárido Man-β1,2-GlcNAc (Yoshida et al., 2001; Vester-Christensen et al., 2013; Endo, 2015, 2019; Sheikh et al., 2017) (Fig. 3). Posteriormente, la enzima α1,6-manosilglicoproteína β1,6-N-acetilglucosaminiltransferasa B (MGAT5B), también conocida como GnT-Vb o GnT-IX, puede catalizar en el complejo de Golgi la adición de una unidad de GlcNAc al núcleo M1 mediante un enlace β1,6 para formar el núcleo M2 (Inamori et al., 2004; Sheikh et al., 2017). Los O-manosilglicanos en los núcleos M1 y M2 son posteriormente elongados de forma secuencial por N-acetilglucosaminiltransferasas (GlcNTs), galactosiltransferasas (GalTs), sialiltransferasas (SiaTs), glucuroniltransferasas (GlcATs), sulfotransferasas (STs) y fucosiltransferasas (FUTs), teóricamente en el complejo de Golgi, aunque los detalles de estas reacciones aún no se conocen (Endo, 2015; 2019) (Fig. 3).
Con respecto a la formación del núcleo M3, el primer paso ocurre en el retículo endoplásmico y consiste en la adición a la O-manosa de una unidad de GlcNAc mediante un enlace β1,4 a partir de una molécula de UDP-GlcNAc, catalizada por la enzima denominada proteína O-manosa β1,4-N-acetilglucosaminiltransferasa 2 (POMGNT2) (Manzini et al., 2012; Endo, 2015, 2019; Sheikh et al., 2017). A continuación, la enzima denominada β1,3-N-acetilgalactosaminiltransferasa 2 (B3GALNT2) cataliza la adición de una unidad de GalNAc mediante un enlace β1,3, con posterior fosforilación en la posición 6 de la O-manosa llevada a cabo por la enzima proteína O-manosa quinasa (POMK) (Yoshida-Moriguchi et al., 2013; Endo, 2015, 2019; Sheikh et al., 2017). Una vez fosforilada la O-manosa, el α-DG viaja al Golgi, donde continúa la elongación del núcleo M3 mediante la actuación de las enzimas fukutina (FKTN) y proteína relacionada con la fukutina (FKRP). Estas proteínas utilizan como sustrato la molécula citidín-difosfato-ribitol (CDP-ribitol), producida por la enzima CDP-L-ribitol pirofosforilasa A (CRPPA), también conocida como ISPD, para sintetizar y añadir dos unidades de ribitol 5-fosfato (Rbo5P) en tándem al núcleo M3 (Kanagawa et al., 2016; Kanagawa y Toda, 2017, 2018).
Finalmente, el núcleo estructural de azúcares M3 se caracteriza por estar elongado en su extremo por una estructura denominada matriglicano, que consiste en un polisacárido compuesto por repeticiones de un disacárido de xilosa (Xyl) y ácido glucurónico (GlcA) (α1,3-Xyl-β1,3-GlcA) y que permite la unión del DG a sus ligandos en la ECM (Yoshida-Moriguchi y Campbell, 2015). La síntesis del matriglicano se inicia mediante la actividad de la enzima ribitol-5-fosfato xilosiltransferasa 1 (RXYLT1), también llamada TMEM5, la cual transfiere la primera molécula de Xyl del matriglicano sobre las unidades de Rbo5P en tándem (Manya et al., 2016; Praissman et al., 2016). A continuación, la enzima β1,4-glucuroniltransferasa 1 (B4GAT1) agrega la primera unidad de GlcA del matriglicano (Praissman et al., 2014; Willer et al., 2014), a partir de la cual la enzima xilosil- y glucuroniltransferasa 1 (LARGE1) o 2 (LARGE2), según el tipo de tejido, elonga la cadena del matriglicano añadiendo repetidamente unidades alternas de Xyl y GlcA (Inamori et al., 2012, 2014; Yoshida-Moriguchi et al., 2013; Praissman et al., 2014; Willer et al., 2014; Endo, 2015, 2019; Yoshida-Moriguchi y Campbell, 2015) (Fig. 3).
DISTROGLICANOPATÍAS
Las distrofias neuromusculares tradicionalmente conocidas como distroglicanopatías (DGPs), clasificadas en la base de datos Online Mendelian Inheritance in Man (OMIM) como distrofias musculares-distroglicanopatías (MDDGs), constituyen un grupo de enfermedades congénitas clínica y genéticamente heterogéneas que se heredan de forma autosómica recesiva y ofrecen en los casos más graves una corta esperanza de vida (Muntoni y Voit, 2004; Reed, 2009). Estos trastornos genéticos pueden derivar de anomalías en la estructura o expresión del propio DG o de una deficiente glicosilación del dominio de tipo mucina del α-DG, lo que tiene como resultado una reducción de su capacidad de unión a sus ligandos de la ECM en tejidos musculares y nerviosos (Martin, 2007). Sus síntomas conllevan un amplio espectro de distrofias musculares congénitas, las cuales aparecen en el momento del nacimiento o en los primeros meses antes de los dos años (edad de adquisición de la marcha), o bien pueden tratarse de distrofias musculares más tardías, como es el caso de las distrofias musculares de cinturas (LGMDs).
Las DGPs se caracterizan por presentar distrofia muscular progresiva acompañada de un aumento (de más de 5-10 veces) en los niveles de creatina quinasa (hiperCKemia) en el músculo, lo cual contribuye a la detección de estas enfermedades (Bönneman et al., 2014). Frecuentemente también se acompañan de anomalías del CNS conducentes a lisencefalia (cerebro liso) en ‘empedrado’, discapacidad intelectual y una serie de defectos oculares que en numerosos casos implican a la retina (Schessl et al. 2006; Reed, 2009; Godfrey et al., 2011).
Las DGPs descritas hasta la fecha, ordenadas de mayor a menor gravedad, son el síndrome de Walker-Warburg (WWS), la enfermedad de músculo-ojo-cerebro (MEB), la distrofia muscular congénita de Fukuyama (FCMD), las distrofias musculares congénitas de severidad intermedia (CMDs) y las LGMDs (Schessl et al. 2006; Reed, 2009; Godfrey et al., 2011; Montanaro y Martin, 2011; Mercuri y Muntoni, 2012). El WWS, la MEB y la FCMD son sindrómicas clínico-neurorradiológicas, mientras que otras CMDs, menos severas a nivel cerebral (incluso con intelecto conservado), así como las LGMDs, generalmente no lo son.
El WWS está considerado como la DGP más severa, y se caracteriza por desarrollarse en edades muy tempranas, es decir, en la fase prenatal o justo después del nacimiento. Los pacientes suelen presentar graves anomalías estructurales del cerebro, como son agiria (ausencia de circunvoluciones cerebrales), lisencefalia severa, hidrocefalia (acumulación excesiva de líquido cefalorraquídeo en el cerebro) y ausencia total o parcial del cuerpo calloso (Beltrán-Valero de Bernabé et al., 2002). También conlleva anomalías oculares, como son cataratas congénitas y microftalmia, y otras que afectan a la retina, como displasia retiniana, atrofia y desprendimiento de la retina, junto a un grave deterioro del desarrollo motor. Los pacientes que padecen este síndrome presentan una esperanza de vida muy corta, de tan solo unos meses (Godfrey et al., 2011).
La MEB y la FCMD muestran una patogénesis similar a la del WWS, aunque con un fenotipo menos grave. Actualmente, el cuadro clínico y radiológico de la MEB que suele asociarse a mutaciones en el gen POMGNT1 se considera equivalente al de la FCMD causada por mutaciones que afectan al gen FKTN, y ofrece una esperanza de vida superior a la de los pacientes de WWS, pudiendo llegar incluso a la edad adulta (Mendell et al., 2006; Dobson et al., 2013). En esta enfermedad es característica la presencia de distrofia muscular congénita, lisencefalia y anomalías estructurales del ojo.
El WWS, la MEB y la FCMD se clasifican como MDDGs de tipo A o MDDGA según el gen mutado que causa la enfermedad: MDDGA1 (OMIM 236670), asociada a mutaciones en el gen POMT1; MDDGA2 (OMIM 613150), en el gen POMT2; MDDGA3 (OMIM 253280), en el gen POMGNT1; MDDGA5 (OMIM 613153), en el gen FKRP; MDDGA6 (OMIM 613154), en el gen LARGE1; MDDGA7 (OMIM 614643), en el gen ISPD (actualmente conocido como CRPPA); MDDGA8 (OMIM 614830), en el gen POMGNT2; MDDGA9 (OMIM 616538), en el gen DAG1; MDDGA10 (OMIM 615041), en el gen RXYLT1; MDDGA11 (OMIM 615181), en el gen B3GALNT2; MDDGA12 (OMIM 615249), en el gen POMK; MDDGA13 (OMIM 615287), en el gen B4GAT1; y MDDGA14 (OMIM 615350), en el gen GMPPB (Tabla 1).
La FCMD o MDDGA4 (OMIM 253800) se debe únicamente a mutaciones en el gen FKTN y es muy frecuente, aunque no exclusiva, en la población japonesa, con una incidencia aproximada de 3/100.000 nacimientos (Ishigaki et al., 2018), debido a una mutación ancestral fundadora originada por la inserción de un retrotransposón de 3 kb en la región 3’-UTR del gen mencionado (Fukuyama et al., 1981; Kobayashi et al., 1998, 2017).
Las CMDs intermedias, o MDDGBs, son DGPs menos severas que las MDDGs de tipo A, aunque también pueden cursar con anomalías cerebrales y oculares. Se caracterizan por ser enfermedades musculares con herencia autosómica recesiva que pueden conllevar o no discapacidad intelectual. Las anomalías musculares no sindrómicas aparecen antes de la edad de adquisición de la marcha en estos pacientes y varían desde miopatía hasta distrofia, dependiendo de la edad a la que se realice la biopsia muscular (Bertini et al., 2011).
Las CMDs han sido clasificadas según el gen asociado como: MDDGB1 (OMIM 613155), causada por mutaciones en el gen POMT1; MDDGB2 (OMIM 613156), en el gen POMT2; MDDGB3 (OMIM 613151), en el gen POMGNT1; MDDGB4 (OMIM 613152), en el gen FKTN; MDDGB5 (OMIM 606612), en el gen FKRP; MDDGB6 (OMIM 608840), en el gen LARGE1; MDDGB14 (OMIM 615351), en el gen GMPPB; y MDDGB15 (OMIM 616992), en el gen DPM3 (Tabla 1). Adicionalmente, cabe mencionar que se han identificado mutaciones en el gen GOSR2 que provocan, además de hipoglicosilación del α-DG y un aumento de los niveles de CK, CMDs que aún no han sido adscritas como MDDG en la base de datos OMIM (Larson et al., 2018) (Tabla 1).
Por último, las LGMDs son las DGPs menos graves y consisten en un grupo heterogéneo de distrofias musculares leves de manifestación tardía (tras la adquisición de la marcha en la edad pediátrica o en fase adulta). Afectan principalmente a las cinturas pélvica escapular, y suelen cursar sin afectación cerebral ni ocular, aunque a veces pueden conllevar cierto grado de de discapacidad intelectual o microcefalia (Godfrey et al., 2011).
Las LGMDs asociadas a defectos en la glicosilación del α-DG se clasifican según el gen responsable como: MDDGC1 (OMIM 609308), causadas por mutaciones en el gen POMT1; MDDGC2 (OMIM 613158), en el gen POMT2; MDDGC3 (OMIM 613157), en el gen POMGNT1; MDDGC4 (OMIM 611588), en el gen FKTN; MDDGC5 (OMIM 607155), en el gen FKRP; MDDGC7 (OMIM 616052), en el gen ISPD; MDDGC8 (OMIM 618135), en el gen POMGNT2; MDDGC9 (OMIM 613818), en el gen DAG1; MDDGC12 (OMIM 616094), en el gen POMK; MDDGC14 (OMIM 615353), en el gen GMPPB; y MDDGC15 (OMIM 612937), en el gen DPM3 (Tabla 1). Adicionalmente, cabe señalar que también se han identificado mutaciones en el gen TRAPPC11 que provocan, además de hipoglicosilación del α-DG y un aumento de los niveles de CK, la LGMD denominada LGMDR18 (OMIM 615356), que aún no ha sido adscrita como MDDG (Larson et al., 2018) (Tabla 1).
Además, existen algunos genes involucrados en la ruta de glicosilación del α-DG asociados a patologías conocidas en conjunto como trastornos congénitos de la glicosilación (CDGs). Estos constituyen una serie de trastornos autosómicos recesivos genéticamente heterogéneos causados por defectos enzimáticos en la síntesis y procesamiento de glicanos u oligosacáridos ligados a asparagina en glicoproteínas (Leroy, 2006). Los CDGs se clasifican según el gen responsable como: CDG1E (OMIM 608799), causados por mutaciones en el gen DPM1; CDG1U (OMIM 615042), en el gen DPM2; y CDG1M (OMIM 610768), en el gen DOLK (Tabla 1).
GENES ASOCIADOS A DISTROGLICANOPATÍAS Y SU FUNCIÓN
Hasta la fecha se han identificado 20 genes asociados a DGPs cuyas mutaciones provocan hipoglicosilación del α-DG, afectando a su función como receptor de ligandos de la ECM. Estos genes codifican, además del propio DG (gen DAG1), un conjunto de glicosiltransferasas conocidas o putativas y otras enzimas, en su mayoría residentes en el retículo endoplásmico o en el Golgi, que están directa o indirectamente implicadas en la adición de O-manosilglicanos al dominio de tipo mucina del α-DG (Bozzi et al., 2009; Endo, 2015, 2019; Ragni et al., 2016; Nickolls y Bönnemann, 2018) y que ya se han citado en este trabajo. En la actualidad se ha descrito la función que desempeñan todos estos genes (si bien en la mayoría de los casos el conocimiento es escaso) y a qué enfermedades concretas (variantes de DGPs) se hallan asociados (Tabla 1), aunque se sigue estudiando la posible implicación de nuevos genes y variantes génicas de los ya conocidos (Johnson et al., 2018; Nickolls y Bönnemann, 2018).
Tabla 1. Genes asociados a distroglicanopatías.
|
Gen |
Proteína |
OMIM |
Variantes de MDDG |
Enfermedades asociadas |
Localización celular |
Referencia principal |
| DAG1 | Distroglicano | 128239 | MDDGA9, C9 | MEB, LGMD | PM, Nuc | Brancaccio, 2019 |
| POMT1 | Proteína O-manosiltransferasa 1 | 607423 | MDDGA1, B1, C1 | WWS, MEB, CMD, LGMD | ER | Godfrey et al., 2007 |
| POMT2 | Proteína O-manosiltransferasa 2 | 607439 | MDDGA2, B2, C2 | WWS, MEB, CMD, LGMD | ER | Godfrey et al., 2007 |
| POMGNT1 | Proteína O-manosa β1,2-N-acetilglucosaminiltransferasa 1 | 606822 | MDDGA3, B3, C3 | WWS, MEB, CMD, LGMD | GC | Mercuri et al., 2009 |
| FKTN | Fukutina | 607440 | MDDGA4, B4, C4 | FCMD, WWS, MEB, CMD, LGMD | GC, Cit, Nuc | Cotarelo et al., 2008 |
| FKRP | Proteína relacionada con la fukutina | 606596 | MDDGA5, B5, C5 | WWS, MEB, CMD, LGMD | ER, GC, Cit, ECM | Brockington et al., 2001 |
| LARGE1 | Xilosil- y glucuroniltransferasa LARGE1 | 603590 | MDDGA6, B6 | WWS, MEB, CMD | GC | Longman et al., 2003 |
| POMGNT2 | Proteína O-manosa β1,4-N-acetilglucosaminiltransferasa 2 | 614828 | MDDGA8, C8 | WWS, LGMD | ER | Manzini et al., 2012 |
| RXYLT1 | Ribitol-5-fosfato xilosiltransferasa 1 | 605862 | MDDGA10 | WWS, MEB | GC | Vuillaumier-Barrot et al., 2012 |
| B3GALNT2 | β1,3-N-acetilgalactosaminiltransferasa 2 | 610194 | MDDGA11 | WWS, MEB | ER, GC | Stevens et al., 2013 |
| POMK | Proteína O-manosa quinasa | 615247 | MDDGA12, C12 | WWS, MEB, LGMD | ER | Di Costanzo et al., 2014 |
| B4GAT1 | β1,4-glucuroniltransferasa 1 | 605517 | MDDGA13 | WWS | GC | Buysse et al., 2013 |
| ISPD | CDP-L-ribitol pirofosforilasa A | 614631 | MDDGA7, C7 | WWS, MEB, LGMD | Cit | Willer et al., 2012 |
| GMPPB | GDP-manosa pirofosforilasa β | 615320 | MDDGA14, B14, C14 | MEB, CMD, LGMD | Cit | Carss et al., 2013 |
| DPM1 | Dolicol-fosfato manosiltransferasa 1 | 603503 | CDG1E | CDG | ER | Kim et al., 2000 |
| DPM2 | Dolicol-fosfato manosiltransferasa 2 | 603564 | CDG1U | CDG | ER | Barone et al., 2012 |
| DPM3 | Dolicol-fosfato manosiltransferasa 3 | 605951 | MDDGB15, C15 | CMD, LGMD | ER | Lefeber et al., 2009 |
| DOLK | Dolicol quinasa | 610746 | CDG1M | CDG | ER | Kranz et al., 2007 |
| TRAPPC11 | Complejo de tráfico de partículas proteicas, subunidad 11 | 614138 | LGMDR18 | LGMD | GC | Larson et al., 2018 |
| GOSR2 | Complejo receptor de SNAP del Golgi, miembro 2 | 604027 | N/A | CMD | GC | Larson et al., 2018 |
Se indican las proteínas codificadas por los 20 genes asociados a DGPs, su número de registro en la base de datos OMIM, las variantes genéticas de distrofias musculares-distroglicanopatías (MDDG) correspondientes, las enfermedades asociadas según la nomenclatura médica tradicional y la localización intracelular de su producto proteico. Abreviaturas: WWS, síndrome de Walker-Warburg; MEB, enfermedad de músculo-ojo-cerebro; FCMD, distrofia muscular congénita de Fukuyama; LGMD, distrofia muscular de cinturas; CMD, distrofia muscular congénita (de severidad intermedia); CDG, trastorno congénito de glicosilación; SNAP, proteína soluble de fijación de factor sensible a N-etilmaleimida; Cit, citosol; Nuc, núcleo; ER, retículo endoplásmico; GC, aparato de Golgi; ECM, matriz extracelular; PM, membrana plasmática; N/A, MDDG aún no adscrita. Tabla elaborada a partir de la base de datos OMIM 2022, Bouchet-Séraphin et al., 2015 y Nickolls y Bönnemann, 2018.
Las DGPs se clasifican en primarias, secundarias o terciarias según su origen genético. Las DGPs primarias están causadas por mutaciones en el gen DAG1, que codifica el propio DG, y son las menos estudiadas, dado que se han detectado muy pocos casos hasta ahora (Hara et al., 2011; Brancaccio, 2019). Las mutaciones en DAG1 ocurren con baja frecuencia y se han identificado en familias consanguíneas, afectando al extremo N-terminal del α-DG. Dichas mutaciones provocan alteraciones estructurales en el DG que comprometen su maduración y dificultan su interacción con las glicosiltransferasas, modificando su estado de glicosilación. También se han encontrado mutaciones que afectan a los extremos C-terminales del α-DG y β-DG, alterando la estabilidad y la unión entre ambas subunidades del DG. Como consecuencia de estas mutaciones, las DGPs primarias originan una serie de síntomas y fenotipos que pueden variar desde distrofia muscular leve con hiperCKemia asintomática hasta LGMD o MEB (Bouchet-Séraphin et al., 2015; Brancaccio, 2019) (Tabla 1).
Las DGPs secundarias, que son las más abundantes, están relacionadas con mutaciones en genes que codifican proteínas directamente implicadas en el proceso de O-glicosilación del α-DG (Bouchet-Séraphin et al., 2015), es decir, glicosiltransferasas de proteínas. Estos genes son: POMT1 y POMT2, que codifican las proteínas que forman el heterocomplejo POMT1/2, el cual cataliza el primer paso de la glicosilación del α-DG añadiendo una unidad de O-manosa a su dominio de tipo mucina; POMGNT1, responsable del primer paso de elongación de O-manosas para la síntesis de los núcleos estructurales M1 y M2 de O-manosilglicanos; los genes POMGNT2, B3GALNT2, POMK, FKTN y FKRP, responsables de la síntesis del núcleo estructural M3; y los que sintetizan la estructura denominada matriglicano sobre el núcleo M3: RXYLT, B4GAT1 y LARGE1. Mutaciones en alguno o varios de estos genes, que codifican glicosiltransferasas, causan DGPs de gravedad variable, desde el WWS hasta LGMDs en orden decreciente de severidad (Lommel et al., 2010; Ragni et al., 2016; Kanagawa y Toda, 2018; Nickolls y Bönnemann, 2018; Endo, 2019) (Tabla 1).
Finalmente, las DGPs terciarias se deben a mutaciones en los genes ISPD, GMPPB, DPM1, DPM2, DPM3, DOLK, TRAPPC11 y GOSR2. Estos genes codifican enzimas responsables de la síntesis de sustratos utilizados en la glicosilación del α-DG, transporte de solutos y partículas proteicas, fijación y recepción de vesículas, etc. Defectos en estas enzimas provocan DGPs que van desde el WWS hasta LGMDs (excepto la FCMD), y CDGs (Brancaccio, 2019) (Tabla 1).
EL DISTROGLICANO EN EL CÁNCER
Durante la progresión del cáncer, el crecimiento normal de las células tumorales primarias se ve descontrolada mediante la modificación de las afinidades de unión de sus receptores en la membrana plasmática. En general, la proliferación e invasión desreguladas van acompañadas en dichas células de alteraciones drásticas en el patrón de expresión de moléculas activas en la interfaz entre la membrana basal de la ECM y la membrana plasmática celular (Sgambato y Brancaccio, 2005; Calogero et al., 2006). En este contexto, se conoce que el DG actúa como un receptor crucial necesario para el desarrollo y mantenimiento de los tejidos epiteliales, musculares y neurales, entre muchos otros (Matsumura et al., 1997; Durbeej et al., 1998; Henry y Campbell, 1998; Bello et al., 2015). La unión de la laminina al α-DG regula la morfología celular mediante la reorganización del esqueleto de actina, modula la expresión génica específica de tejido y promueve la supervivencia y diferenciación celulares sobre la proliferación y migración. A medida que se produce la pérdida del DG, todas estas funciones se ven afectadas durante la progresión del tumor en una lista creciente de neoplasias humanas y líneas celulares derivadas de tumores (Matsumura et al., 1997; Muschler et al., 2002; Weir et al., 2006; Beltrán-Valero de Bernabé et al., 2009).
Se han detectado deficiencias en la expresión y, más frecuentemente, en la glicosilación del DG mediante el uso de anticuerpos que reconocen como epítopos estructuras específicas de carbohidratos O-manosilados en el α-DG que forman parte del matriglicano. Dichos anticuerpos, como son IIH6 y VIA4-1, se han utilizado tanto en análisis de Western blotting como de inmunocito/histoquímica. Estas alteraciones se han detectado en una amplia variedad de tumores y líneas celulares, asociadas a la tumorigénesis, a la que contribuye la pérdida de adhesión de las células a la ECM (Moore y Winder, 2010; Montanaro y Martin, 2011). Adicionalmente, la subunidad β del DG es una proteína versátil que actúa como receptor para la interacción célula-ECM y como una plataforma multifuncional para la remodelación del citoesqueleto, la dinámica de adhesión celular, la transducción intracelular de señales y la modulación de la estructura y actividad nucleares (Moore y Winder, 2010; Vélez-Aguilera et al., 2018). Alteraciones en estos procesos moduladores relacionados con el DG se están detectando en una variedad creciente de células tumorales humanas, especialmente en cánceres derivados de tejidos epiteliales.
CONCLUSIONES Y PERSPECTIVAS
El DG es una glicoproteína localizada en la membrana plasmática formando parte del DGC, que es esencial para la unión de determinadas proteínas de la ECM a la actina del citoesqueleto a través de la distrofina en células del músculo esquelético y tejidos no musculares. Actualmente se conocen un total de 20 genes implicados en la correcta expresión y glicosilación del DG cuyas mutaciones afectan a su función como receptor de ligandos de la ECM, provocando una serie de distrofias neuromusculares congénitas conocidas como DGPs. Entre estos genes se encuentra DAG1, que codifica el propio DG, y un conjunto de genes que codifican glicosiltransferasas conocidas o putativas así como otras enzimas, en su mayoría residentes en el retículo endoplásmico o en el Golgi, que están directa o indirectamente implicadas en la O-manosilglicosilación del dominio de tipo mucina del α-DG. La dilucidación de la función que desempeñan todos estos genes, que en la mayoría de los casos es poco conocida, y de las variantes concretas de DGPs a las que se hallan asociados, es esencial para mejorar la comprensión de su implicación en la ruta de glicosilación del DG y la importancia de su papel en cada tejido. También es importante el estudio de la posible implicación en esta ruta de nuevos genes y variantes génicas de los ya conocidos.
Adicionalmente, alteraciones en la expresión y/o glicosilación del DG se han descrito con frecuencia en una amplia variedad de distintos tipos de tumores humanos y líneas celulares derivadas de cáncer. Se ha detectado una correlación entre la hipoglicosilación del α-DG y su pérdida de unión a laminina y, consecuentemente, del anclaje celular a la ECM del tejido en el que se origina el tumor. Asimismo, alteraciones en el papel del β-DG como molécula señalizadora y moduladora indirecta de la expresión génica se han relacionado con el contexto de la patogénesis del cáncer. Por tanto, las deficiencias en la expresión y/o glicosilación del DG se postulan como un hecho relevante que contribuye a la tumorigénesis y malignidad del cáncer, conduciendo probablemente a un aumento en el crecimiento y la proliferación celular, junto a la adquisición de propiedades de migración e invasividad, y derivando finalmente en un mal pronóstico y metástasis.
Por todo ello, se puede concluir que el estudio del DG y de genes y proteínas asociados a DGPs sigue siendo de suma importancia, no solo para la comprensión de las DGPs y el diseño de mejores tratamientos orientados a los pacientes que sufren estas distrofias neuromusculares, sino también para esclarecer el posible papel preventivo del DG sobre el desprendimiento celular de la ECM, la división celular descontrolada y la invasividad celular. Esto último debe proporcionar un marco de trabajo que permita identificar nuevos biomarcadores moleculares de diagnóstico y pronóstico del cáncer, así como nuevas dianas terapéuticas objeto de tratamientos más especializados.
AGRADECIMIENTOS
La investigación de los autores está financiada por la Federación Española para la Ciencia y la Tecnología (FECYT) crowdfunding ref. PR320, por el Rotary Club Torrevieja donación ref. 1-19XPA, y por las ayudas de la Universidad de Alicante ref. VIGROB21-237, UADIF21-77 y UAUSTI21-12 a JMN.
Declaración de ausencia de conflictos de intereses
Los autores declaran que no tienen ningún conflicto de intereses con el contenido de este artículo.


