INTRODUCCIÓN
El síndrome de Temple o disomía uniparental materna 14 (Bertini et al., 2017) (OMIM #616222) es un trastorno por impronta, al igual que los síndromes de Angelman (OMIM #105830) o de Prader-Willi (OMIM #176270). La impronta es un proceso biológico por el cual la expresión de ciertos genes depende del origen parental, de tal forma que la copia materna o paterna se inactiva de una forma programada epigenéticamente (Beygo et al., 2018). Esta expresión diferencial puede ser específica de tejidos (como en el caso del síndrome de Angelman, asociado a alteraciones en el gen UBE3A) o del desarrollo (Camprubi-Sánchez et al., 2006). Además, los genes sometidos a impronta tienden a agruparse en regiones cromosómicas concretas, como en 14q32. Las enfermedades de impronta pueden estar causadas por varios mecanismos: mutación genética, deleción o duplicación cromosómica, disomía uniparental y defectos de impronta. A excepción de las mutaciones en el ADN de un gen improntado, el resto de los mecanismos asocian patrón alterado de metilación del ADN y no son detectables mediante estudios de secuenciación. Se estima que el 1% de los genes humanos están sometidos a impronta, sin embargo, solo se han identificado trece enfermedades de impronta (Serra-Juhe y Pérez-Jurado, 2015) (ver tabla 1).
Tabla 1: Enfermedades de la impronta (Serra-Juhe y Pérez-Jurado, 2015).
| Enfermedades de la impronta | Localización citogenética | Nomenclatura OMIM |
| Diabetes neonatal transitoria asociada a 6q24 | (6q24) | (TNDM, OMIM#601410) |
| Síndrome Beckwith-Wiedemann | (11p15) | (BWS, OMIM#130650) |
| Síndrome Silver-Russell | (11p15) | (SRS, OMIM#180860) |
| Síndrome Temple | (14q32) | (síndrome UPD 14 materna) (OMIM#616222) |
| Síndrome Kagami-Ogata | (14q32) | síndrome (UPD 14 paterna) (OMIM#608149) |
| Síndrome Angelman | (15q11-13) | (AS, OMIM#105830) |
| Síndrome Prader-Willi | (15q11-13) | (PWS, OMIM#176270) |
| Pseudohipoparatiroidismo tipo 1B | (20q13) | (PHP1B, OMIM#603233) |
| Disomía materna del cromosoma 20 | (20q11-q13) | (MBCS, #617352) |
| Pubertad precoz central | (15q11, gen MKRN3) | (PPCB2, OMIM#615346) |
| Síndrome de Birk Barel | (8q24, gen KCNK9) | (OMIM#612292) |
| Síndrome IMAGe | (11p15.5, gen CDKN1C) | (OMIM#614732) |
| Mola hidatiforme recurrente familiar | (19q13.4, gen NLRP7) | (MHR, OMIM#231090) |
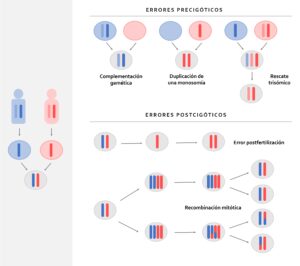
El síndrome de Temple es una entidad de muy baja prevalencia y poco conocida, debido a la pérdida funcional de los genes de expresión paterna en 14q32, que puede estar causada por deleción, por pérdida de impronta paterna o por disomía uniparental materna. Las tres situaciones asocian una pérdida de la metilación del gen GTL2, metilado en el cromosoma paterno (Gillessen-Kaesbach et al., 2018) (Figura 1). Este locus abarca genes codificadores de proteínas expresados paternamente (DLK1, RTL1 y DIO3) y lncRNA expresados maternamente (MEG3/GTL2, RTL1as y MEG8), así como numerosos miRNA y snoRNA. El control de la expresión es complejo, con tres regiones metiladas diferencialmente que regulan la transcripción de línea germinal, placentaria y específica de tejido (Prasasya et al., 2020). El fenotipo clínico es muy similar al descrito para el síndrome de Prader-Willi, con retraso del crecimiento pre y postnatal, hipotonía neonatal, dificultades en la alimentación en la primera infancia con escasa ganancia ponderal, talla baja, anomalías óseas variables (manos y pies pequeños, dolicospondilia, huesos largos y delgados y desproporción craneofacial), dismorfia facial leve (frente ancha y prominente, paladar ojival, micrognatia y nariz corta con raíz plana y punta ancha) y retraso en el desarrollo con o sin discapacidad intelectual leve (Prasasya et al., 2020; Lande et al., 2018). Con el tiempo suelen presentar obesidad central y pubertad prematura. Descrito por primera vez por Temple y colaboradores en 1991, actualmente hay reportados menos de 100 casos, con una frecuencia estimada de <1/1.000.000. Sin embargo, varios autores señalan la posibilidad de que esté infradiagnosticada (Beygo et al., 2018) y por tanto la necesidad de incluirla dentro de la búsqueda etiológica de los pacientes con fenotipo Prader-Willi o en lactante hipotónico (Mesquita et al., 2018).
Aquí se describen los hallazgos clínicos y epigenéticos de un paciente con síndrome de Temple concebido mediante fecundación in vitro, y se discute la utilidad del diagnóstico precoz en el pronóstico y manejo clínico del síndrome.
CASO CLÍNICO
Aportamos un caso de un lactante diagnosticado de forma precoz en contexto del estudio genético por sospecha inicial de síndrome de Prader-Willi. Se trata de un varón, fruto de un embarazo gemelar bicorial biamniótico concebido mediante fecundación in vitro con ovodonación. En la semana 29 de gestación se detectó un retraso del crecimiento intrauterino. Fue prematuro de 35 semanas con peso de 1320g (p<3). Desde el nacimiento se detectó importante hipotonía y retrognatia. No presentó alteraciones en la alimentación. A la edad de 15 meses, el paciente presenta retraso en todas las áreas del desarrollo, especialmente a nivel motor. En la exploración presenta hipotonía grave e hiperlaxitud ligamentosa. Presenta un fallo de medro muy significativo con peso en percentil <3 (-4,88 DE). Además, destacan los rasgos dismórficos leves con frente ancha y levemente abombada (percentil de perímetro cefálico normal), estrabismo ocular, nariz con raíz nasal aplanada, comisuras de la boca hacia abajo y paladar ojival muy pronunciado. Estas anomalías a nivel orofacial repercuten significativamente en el proceso de alimentación, con importante dificultad en la masticación y deglución.
A la edad de dos meses, el estudio mediante RMN cerebral objetivó quiste aracnoideo de pequeño tamaño, sin otras alteraciones. También se descartó CMV congénito, los potenciales evocados auditivos fueron normales, al igual que el estudio del electromiograma. A esta edad se realizó estudio genético para el síndrome de Prader-Willi, en el que se descartó alteración del patrón de metilación en el locus 15q11.2-q13, pero se detectó pérdida del patrón de impronta paterno en 14q32 por epimutación (Mayo et al., 2015).
DISCUSIÓN
El Síndrome de Temple es una enfermedad genética desconocida para la mayor parte de los clínicos, en parte debido a su baja prevalencia. Dadas las similitudes clínicas con el síndrome de Prader-Willi, especialmente en época neonatal, en nuestro centro se realiza de forma sistemática el estudio de metilación de ambos síndromes tal como se ha descrito previamente (Mayo et al., 2015). Esto nos ha permitido diagnosticar seis pacientes distintos en los últimos años, lo que sugiere que éste no es un síndrome tan infrecuente. Clínicamente comparte características con el Prader-Willi, aunque con mejor pronóstico en su desarrollo cognitivo, dato de relevancia en lactantes. Hay evidencia de una leve reducción de la capacidad intelectual (CI medio 75-95) (Gillessen-Kaesbach, et al., 2018, Ioannides et al., 2014), que por otra parte podría estar relacionada con el hecho de que un 30% de estos pacientes son prematuros y presentan dificultades en la alimentación en la primera infancia. También se ha postulado que puede haber un sesgo y que sean más diagnosticados y/o evaluados aquellos pacientes más afectados. Con la evolución, aproximadamente la mitad de los casos desarrollan obesidad central, con mayor incidencia de diabetes tipo 2 y riesgo cardiovascular global (Ioannides et al., 2014).
Ante un paciente con este fenotipo y síndrome de Prader-Willi descartado, se debe de completar estudio genético buscando dicha alteración. Estos pacientes requieren seguimiento multidisciplinar, por parte de neuropediatría, rehabilitación, oftalmología y endocrinología, sin olvidar la importancia del asesoramiento genético familiar. Pensamos que un correcto seguimiento y tratamiento, puede mejorar la evolución y calidad de vida de estos pacientes. En este sentido, se ha descrito que estos pacientes pueden beneficiarse del tratamiento con hormona de crecimiento (GH) por lo que habría que valorar instaurar tratamiento en todos estos pacientes (Brightman et al., 2018).
Significativamente, se ha descrito que los niños nacidos por técnicas de reproducción asistida presentan mayor riesgo de alteraciones de la impronta, especialmente para los síndromes de Angelman y Beckwith-Wiedemann (Odom y Segars, 2010; Vermeiden y Bernardus, 2013). Este es un tema controvertido, donde queda por dilucidar si las alteraciones de la impronta están causadas por las técnicas de reproducción asistida en sí, o bien si podrían ser consecuencia del defecto de fertilidad subyacente (Sciorio y Esteves, 2022). Sea cual sea la relación de causalidad, de momento esta asociación no se ha descrito con el síndrome de Temple, siendo este el primer caso del que tenemos conocimiento.
Agradecimientos
Los autores desean agradecer a la familia por participar en este estudio y al equipo de la Unidad de Genómica del Hospital La Fe.
Financiación y conflictos de interés
Contrato Río Hortega CM19/00181
No existen conflictos de interés.