
Los sentidos de la vista, el equilibrio y el oído son esenciales para todas las actividades importantes de la vida diaria, desde las interacciones sociales y la movilidad hasta la apreciación de la música, el arte y la naturaleza. En consecuencia, el deterioro de estas modalidades sensoriales tiene un profundo impacto negativo en la calidad de vida de las personas afectadas, que van desde limitaciones de comunicación a menor acceso a entretenimiento y oportunidades laborales. A su vez, esto puede conducir al aislamiento social y la depresión (Bonnet y El-Amraoui, 2012; Mathur y Yang , 2019; Delmaghani y El-Amraoui, 2022). A partir de datos empíricos completos, la Organización Mundial de la Salud estima que alrededor de 285 millones de personas actualmente presentan deficiencias visuales graves en todo el mundo (WHO, 2021). Además, 460 millones de personas, que representan el 5% de la población mundial, tienen una discapacidad auditiva incapacitante (con o sin déficit de equilibrio). Se espera que este número aumente a más de mil millones de personas para 2050 (WHO, 2021). En conjunto, estos déficits sensoriales tienen un impacto económico dramático en los sistemas de salud y la sociedad en su conjunto.
Como dijo Helen Keller, la primera persona sordociega en obtener un título universitario, “La ceguera separa a las personas de las cosas; la sordera separa a las personas de las personas”, señalando la singularidad y los desafíos de ser sordociego.
EL SÍNDROME DE USHER
El síndrome de Usher (USH), es la causa más frecuente de sordoceguera hereditaria en humanos, siendo responsable de más del 50% de los individuos sordociegos. También está implicado en un 5% de todas las sorderas congénitas y en un 18% de los casos de retinosis pigmentaria (RP). El USH consiste en la asociación de sordera, una distrofia progresiva de la retina conocida como RP y, en ocasiones, alteración del equilibrio. En España la prevalencia se estima en un 4,2/100.000 nacidos vivos (Espinós et al., 1998).
Históricamente, a nivel clínico se han definido tres tipos de USH (USH1 a 3), con alrededor de 10 genes causales identificados hasta el momento. Estos tres tipos son principalmente clasificados según la gravedad y progresión de la hipoacusia y la presencia o ausencia de alteración vestibular (Tabla 1). Sin embargo, hay algunos pacientes en los que las manifestaciones clínicas no encajan exactamente con las características de los tres grupos definidos, clasificándose como síndrome de Usher atípico (Fuster-García et al., 2021).
| Síndrome de Usher tipo 1 | Síndrome de Usher tipo 2 | Síndrome de Usher tipo 3 | |
|
Pérdida auditiva |
Severa a profunda | Moderada a severa | Variable |
| Congénita | Congénita | Postlingual | |
| Estable | Estable | Progresiva | |
| Inicio de la RP | Antes de la pubertad | Después de la pubertad | Variable |
| Función vestibular | Arreflexia vestibular | Normal | Variable |
Tabla 1. Clasificación clínica del Síndrome de Usher.
[row]
[col span__sm=»12″ padding=»0px 20px 0px 20px» margin=»0px 0px 0px 0px» bg_color=»#e6e6e6″ bg_radius=»3″ depth=»1″ depth_hover=»1″]
Síndrome de Usher tipo 1
El síndrome de Usher tipo 1 (USH1) es la forma más grave de la enfermedad y está caracterizado por hipoacusia neurosensorial congénita, de grave a profunda. Estos pacientes pierden los restos de audición a lo largo de su primer año de vida, de forma que no serán capaces de desarrollar el habla, a menos que sean tratados con un implante coclear.
Además, el USH1 se caracteriza por una hiporreflexia vestibular. La afectación del aparato vestibular se refleja ya en el primer año de vida conduciendo a un retraso en el desarrollo motor.
En los pacientes USH1 los primeros signos de ceguera nocturna aparecen en la primera década de vida (Fuster-García et al., 2021).
Síndrome de Usher tipo 2
El síndrome de Usher tipo 2 (USH2) es el tipo clínico más frecuente y, en general, menos grave que el USH1. El grado de hipoacusia es leve o moderada en frecuencias bajas, a grave en frecuencias más elevadas, tendiendo a permanecer estable. Aunque la pérdida auditiva es congénita, los niños son capaces de desarrollar el habla y poder comunicarse. Además, el grado de audición mejora notablemente mediante el uso de audiófonos. La función vestibular es normal, por lo que los niños no sufren alteraciones en su desarrollo motor.
Los síntomas de la RP se manifiestan habitualmente de forma más tardía en los USH2, generalmente en la segunda década de vida. Sin embargo, la edad de inicio de la RP puede variar enormemente dentro de los USH1 y USH2, pudiendo haber solapamientos entre ambos tipos. El rango y grado de la pérdida visual es muy variable inter e intrafamiliarmente, pero generalmente tiende a progresar de forma más lenta que los USH1 (Stemerdink et al., 2022).
Síndrome de Usher tipo 3
Es el tipo USH menos frecuente con diferencia, excepto en poblaciones endogámicas como la población finlandesa o los judíos askenazi. Los pacientes con síndrome de Usher tipo 3 (USH3), sufren una hipoacusia neurosensorial postlingual y progresiva, pudiendo aparecer desde la primera a la tercera década de vida. En etapas iniciales el grado de pérdida auditiva es similar al USH2, con un mayor impacto en las frecuencias elevadas. La progresión es muy variable, pero en la mayor parte de los casos se llega a una sordera profunda alrededor de la cuarta década de vida. Los niveles de audición permanecen generalmente normales durante la etapa de adquisición del lenguaje, de forma que estos niños no tienen problemas para desarrollar el habla. La presencia de alteración en la función vestibular es variable, encontrándose en un 50% de los pacientes USH3 (Marouf et al., 2022).
[/col]
[/row]
GENES Y PROTEÍNAS IMPLICADOS EN EL SÍNDROME DE USHER
El síndrome de Usher es heterogéneo clínicamente, pero también a nivel genético, ya que cada tipo clínico puede a su vez dividirse en subtipos genéticos. Hasta el momento se han descrito seis genes para el USH1, tres para el USH2 y un único gen para el USH3 (Tabla 2).
Además, se ha descrito la asociación del gen PDZD7 en el síndrome de Usher, bien en un modelo de digenismo junto al gen ADGRV1 o como modificador, agravando el fenotipo retiniano en pacientes con mutaciones en USH2A.
La publicación de estudios sobre la localización e interacciones de las proteínas codificadas por los genes Usher ha puesto en evidencia la existencia de una red proteica constituida por estas proteínas, conocida como “interactoma Usher”. Esta red la encontraríamos tanto en la retina como en el oído interno, donde desempeñan un papel esencial para el correcto funcionamiento de ambos sistemas (Reiners et al., 2006).
| Tipo clínico | Locus | Localización cromosómica | Gen | Proteína |
| USH1 | USH1B | 11q13.5 | MYO7A | miosina VIIa |
| USH1C | 11p15.1 | USH1C | harmonina | |
| USH1D | 10q22.1 | CDH23 | cadherina 23 | |
| USH1F | 10q21.1 | PCDH15 | protocadherina 15 | |
| USH1G | 17q25.1 | USH1G | SANS | |
| USH1J | 15q24 | CIB2 | calcium and integrin binding protein 2 | |
| USH2 | USH2A | 1q41 | USH2A | usherina |
| USH2C | 5q14.3 | ADGRV1 | Adhesion G Protein-Coupled Receptor V1 | |
| USH2D | 9q32 | WHRN | whirlina | |
| USH3 | USH3A | 3q25.1 | CLRN1 | clarina 1 |
Tabla 2. Loci, genes y proteínas identificados en el síndrome de Usher.
DIAGNÓSTICO MOLECULAR DEL SÍNDROME DE USHER
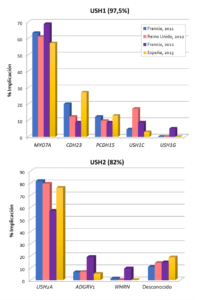
La secuenciación de nueva generación (NGS del inglés Next Generation Sequencing) ha supuesto una revolución en el diagnóstico de las enfermedades genéticamente heterogéneas. La aplicación de un panel que incluye los 10 genes responsables de síndrome de Usher ha permitido diagnosticar genéticamente a casi el 100% de los casos con síndrome de Usher tipo 1 y más del 80% de los casos tipo 2.
Entre los casos tipo 1, el gen más frecuente es MYO7A que cuenta para más del 50% de los casos, le siguen en prevalencia, CDH23 y PCDH15. Para el tipo 2, el gen USH2A está implicado en casi el 80% de los casos. Esta distribución por genes es muy similar en todas las poblaciones europeas estudiadas (Figura 1). Sin embargo, secuenciando todos los genes conocidos, hay casos que permanecen sin diagnosticar, especialmente para el USH2 (Bonnet et al., 2011).
La secuenciación de exomas y genomas completos ha permitido identificar genes que dan lugar a fenotipos muy similares al síndrome de Usher (Usher-like) como los genes CEP250, CEP78, ESPN y otros que pueden explicar un porcentaje de los casos no diagnosticados genéticamente. También se han encontrado casos en los que una misma persona era portadora de mutaciones en un gen de sordera y un gen de RP dando lugar a un fenotipo de USH.
TRATAMIENTOS PARA EL SÍNDROME DE USHER
El síndrome de Usher afecta principalmente a los órganos de la visión y la audición. Actualmente, no existe ninguna cura para el síndrome de Usher. Sin embargo, se están desarrollando distintos tipos de estrategias para mejorar la calidad de vida de los pacientes que la padecen.
Tratamiento contra la hipoacusia
En la hipoacusia neurosensorial el órgano afectado es el órgano de Corti, situado en el oído interno. Dependiendo el grado de hipoacusia que presente el paciente, existen diferentes tipos de estrategias:
- Audífonos: se suele usar en pacientes que padecen sorderas de leve a moderada (USH2). Este aparato electrónico permite la amplificación de los sonidos.
- Implantes cocleares: se suelen utilizar en pacientes que presentan una sordera congénita profunda (USH1) o progresiva que deriva en profunda (USH3). Es un pequeño dispositivo electrónico que se implanta mediante cirugía y sustituye de manera artificial al órgano de Corti (Loundon et al., 2003).
Perspectivas de terapia para la retinosis pigmentaria
La retinosis es una degeneración progresiva, para la que todavía no existe tratamiento a excepción de una terapia génica ya comercializada para pacientes con mutaciones bialélicas en el gen RPE65 (Miraldi et al., 2018). Sin embargo, las características anatómicas de la retina como son su accesibilidad y su barrera interna, que limitan la difusión de sustancias o vectores al resto del organismo, hacen que los avances en la terapia contra esta enfermedad sean muy prometedores.
Dependiendo el estadio de la enfermedad donde nos encontremos, las estrategias aplicables son distintas. En estadios tempranos, se puede llevar a cabo tanto la aplicación de factores que ralenticen el proceso de degeneración, como son los factores neurotróficos, antinflamatorios o que eviten el estrés oxidativo. En fases intermedias de la enfermedad en las que aún quedan fotorreceptores se podría utilizar la terapia génica. En etapas finales, únicamente se podría aplicar los trasplantes de retina o los implantes retinianos debido al deterioro que presentan los fotorreceptores.
Terapias farmacológicas
Se están ensayando moléculas neuroprotectoras como algunos factores neurotróficos que protegen a los fotorreceptores de los procesos de muerte celular que desencadena la RP. También se están ensayando fármacos que retrasan la degeneración de la retina como la minociclina, mioricina, dexametasona, anti-TNFα o el estabilizador de HIF-1α DMOG. Estas terapias están dirigidas a tratar la RP independientemente de la mutación responsable (Olivares-González et al., 2021). Por último, se ha ensayado un fármaco, PTC124, que es específico para mutaciones que dan lugar a una proteína truncada (Goldmann et al., 2011).
Terapia génica
Se basan en la sustitución o reparación del gen que presenta la mutación, por lo tanto, es necesario conocer cuál es el gen mutado en cada paciente. Como se ha dicho previamente, se están realizando estudios preclínicos mediante varias aproximaciones, reemplazamiento del gen mutado por uno normal, oligonucléotidos antisentido o edición génica mediante CRISPR (Fuster-García et al., 2017).
Recientemente, se ha llevado a cabo un ensayo clínico con un lentivirus que contiene el gen MYO7A denominado SAR421869 en pacientes con mutaciones en dicho gen. Este ensayo se abandonó en septiembre de 2019, no por problemas de bioseguridad, sino porque el patrocinador del estudio decidió no continuar con el desarrollo del producto. Ha comenzado otro para determinar la seguridad a largo plazo, tolerabilidad y actividad biológica del UshStat, con características similares al anterior, también para pacientes con mutaciones en MYO7A.
En 2018 comenzó un proyecto del programa Horizonte 2020 de la Unión Europea en el que participan la Fundación Jiménez Díaz de Madrid y el Hospital La Fe de Valencia y en el que se ensayará un tipo de terapia que se denomina terapia génica dual también para pacientes con mutaciones en el gen MYO7A. Este estudio sigue en marcha.
Lamentablemente, no todos los ensayos llegan a buen puerto y en agosto de 2022 se abandonó un ensayo clínico en fase III basado en la tecnología de oligonucleótidos antisentido para pacientes con síndrome de Usher tipo 2 y con mutaciones en el exón 13 del gen USH2A ya que no se alcanzaron los resultados esperados.
Terapia celular
No existe una terapia celular específica para el síndrome de Usher, pero sí hay estudios donde se usa la terapia celular como aproximación al tratamiento de las distrofias de retina. Hay varios ensayos clínicos en marcha con células mesenquimales de médula ósea, de células troncales de cordón umbilical para la RP o de células embrionarias humanas derivadas a epitelio pigmentario de la retina e incluso a fotorreceptores (Mellough et al., 2014). Los principales problemas para este tipo de terapia son encontrar un linaje celular o un tejido a partir del cual se puedan derivar las células que se desea reponer y los problemas que conllevan tanto la desdiferenciación como la reprogramación de estas células troncales y la funcionalidad de las mismas.
Prótesis de retina
En las etapas finales de las DR apenas queda resto visual y ninguna de las terapias anteriores es útil. En estos casos, se está experimentando con cierto tipo de prótesis artificiales, cámaras que conectan directamente con microchips electrónicos implantados en la retina y que permiten al menos la percepción de luz y la distinción de algunas formas (Luo & da Cruz, 2016).
Conflictos de interés
No existen conflictos de interés.


