
Las enfermedades neurodegenerativas con acumulación cerebral de hierro (ENACH; NBIA, neurodegeneration with brain iron accumulation) comprenden un grupo de enfermedades raras caracterizadas por la acumulación anormal de hierro en los ganglios basales. Se estima que su prevalencia es de 1-3 personas cada 1.000.000. En cuanto a las manifestaciones clínicas, se caracteriza por progresivos trastornos del movimiento derivados de afectaciones extrapiramidales, como son el parkinsonismo, la distonía y la corea. Alteraciones neuropsiquiátricas y discapacidad intelectual también se incluyen entre los aspectos más característicos de este grupo de enfermedades.
Hasta la fecha, se han aceptado diez formas NBIA causadas por mutaciones en genes implicados en diversas rutas biológicas, como son el metabolismo del hierro o de los lípidos, la dinámica mitocondrial o la autofagia: PANK2, PLA2G6, WDR45, C19ORF12, FA2H, ATP13A2, COASY, FTL1, CP y DCAF17.
Tanto la edad de inicio de los síntomas como la esperanza de vida tras la aparición de éstos son variables, en función de la forma NBIA presente. Generalmente, se tratan de casos pediátricos con una esperanza de vida limitada, perdiendo la capacidad deambulatoria normalmente tras 10-15 años desde el diagnóstico.
Las dos formas NBIA mayoritarias son PKAN (PANK2-associated neurodegeneration; 30-50% de los casos) causada por mutaciones en el gen PANK2, y PLAN (PLA2G6-associated neurodegeneration; 20%) como consecuencia de mutaciones en el gen PLA2G6. Las siguientes en la lista serían MPAN (6-10%) que debido a mutaciones en el gen C19ORF12 y BPAN (1-2%) asociada a variantes en el gen WDR45. El resto de formas son extremadamente raras y los casos descritos son escasos. Sin embargo, aun estudiando todos estos genes, un número relevante de pacientes NBIA permanecen sin un diagnóstico genético concluyente, lo que remarca la elevada heterogeneidad genética en estas enfermedades y, por otra parte, nos indica que existen otros genes NBIA todavía no conocidos.
Más allá de los signos clínicos predominantes, las formas NBIA cursan con expresividad variable. En algunos casos, además, es complejo determinar si la acumulación cerebral de hierro es causa o consecuencia de la patología, dificultando la discriminación de aquellos pacientes NBIA de los que cursan con otras alteraciones metabólicas que también conducen a una acumulación de hierro anómala como un efecto secundario debido a la neurodegeneración.
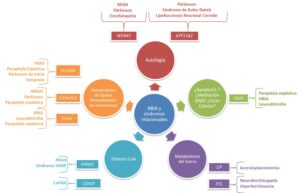
Ya que el cerebro es especialmente sensible a las especies reactivas del oxígeno (ROS, reactive oxygen species) debido a su alto consumo de este elemento, sus limitados compuestos antioxidantes y la concentración alta de iones metálicos, las diez formas NBIA establecidas están vinculadas a eventos de disfunción mitocondrial, estrés oxidativo y neuroinflamación, a través de la alteración de distintas rutas fisiológicas (Hinarejos et al., 2020):
-
Biosíntesis de CoA
Mutaciones en PANK2 y CoPAN conducen a trastornos que presentan alterada la ruta de biosíntesis de la coenzima A (coA).
PANK2 codifica la isoforma mitocondrial de la enzima pantotenato-quinasa, esencial en la regulación del primer paso de la ruta de síntesis de CoA. Se conocen más de 120 mutaciones asociadas a PKAN cuya herencia es autosómica recesiva (AR). La presentación clásica se da en la infancia, aunque se han descrito formas atípicas en edad adulta, y cursa con distonía, parkinsonismo, espasticidad, retinosis pigmentaria, acantocitosis, alteraciones cognitivas y neuropsiquiátricas. En neuroimagen, el PKAN es característica por el “ojo del tigre”, que consiste en un foco hiperintenso central en el globo pálido con una área periférica hipointensa, como consecuencia de los depósitos de hierro en esta región.
COASY codifica una enzima bifuncional que cataliza los 2 últimos pasos de la biosíntesis de CoA. Mutaciones en este gen son responsables de la forma CoPAN (COASY-protein associated neurodegeneration) con herencia AR. Han sido descritas cuatro familias y tres variantes de cambio de aminoácido en relación con esta enfermedad. La manifestación clínica es temprana y se caracteriza por alteración de la marcha, dificultades en el aprendizaje, distonía y espasticidad. También se detecta hipointensidad periférica con hiperintensidad central en el globo pálido, en neuroimagen.
-
Metabolismo de lípidos y remodelación de membranas
Mutaciones en los genes PLA2G6, c19orf12 y FAHN causan formas NBIA que dan lugar a defectos en el metabolismo de los lípidos y la estabilidad de las membranas, todas ellas con herencia AR
PLA2G6 codifica distintas isoformas de la fosfolipasa independiente de calcio A2 (grupo VI), implicada en la homeostasis de fosfolípidos, y mutaciones en este gen dan lugar a la forma PLAN. Más de 200 mutaciones en PLA2G6 han sido descritas. Las formas clásicas comienzan en la infancia y se caracterizan por un deterioro progresivo de las capacidades motoras y cognitivas, hipotonía troncal, atrofia óptica, signos piramidales y espasmos. Formas atípicas comienzan en la adolescencia o principio de la edad adulta y, en estos casos, es característica la manifestación de distonía-parkinsonismo.
C19orf12 codifica una proteína mitocondrial vinculada a la homeostasis de lípidos, pero su función es desconocida. Mutaciones en este gen dan lugar a la forma MPAN (mitocondrial-membrane protein-asociated neurodegeneration), caracterizada por una marcada neuropatía de inicio tanto en la infancia como al comienzo de la edad adulta. En torno a 50 mutaciones han sido descritas en C19orf12. Las manifestaciones clínicas incluyen para- o tetraparesia espástica y atrofia muscular, siendo distonía y parkinsonismo también frecuentes. En cuanto al diagnóstico por imagen, en estos pacientes se detecta hipointensidad en el globo pálido y la sustancia negra, además de hiperintensidad en globo pálido.
FA2H codifica una enzima mono-oxigenasa NADPH-dependiente, crucial en el neurodesarrollo, participando en la formación de mielina. Se han descrito en torno a 65 variantes en FA2H que causan FAHN (fatty acid hydroxylase-associated neurodegeneration). El inicio de los síntomas es temprano, y los pacientes cursan con alteración de la marcha, paraplegia espástica, ataxia y distonía.
-
Regulación del autofagosoma/lisosoma
Mutaciones en WDR45 y ATP13A2 causan formas NBIA en las que las rutas de autofagia y la vía lisosomal están alteradas.
WDR45 codifica un miembro de la familia de proteínas WD40, implicadas en interacciones proteína-proteína, autofagia, ciclo celular y control transcripcional. WDR45 es esencial para la formación del autofagosoma. Mutaciones en este gen causan la forma BPAN, que sigue una herencia dominante ligada al cromosoma X. Hasta la fecha, alrededor de 100 variantes en este gen han sido descritas, y todas ellas en casos esporádicos (mutaciones de novo) mayoritariamente mujeres, lo que indicaría que para hombres pudiera ser letal antes del nacimiento. En la infancia, la sintomatología se asemejaría al síndrome de Rett, mientras que en adultos es característica un síndrome distonía-parkinsonismo, la demencia, los espasmos y la ataxia. En pacientes BPAN se detecta hipointensidad del globo pálido y la sustancia negra, con línea central hiperintensa, en neuroimagen.
ATP13A2 codifica una ATPasa lisosomal de tipo 5P, localizada en endosomas, lisosomas y autofagosomas. Participa en la homeostasis del manganeso, el zinc y el hierro, por lo que está implicado en su transporte, en la bioenergética mitocondrial y en la ruta autofágica/lisosomal. Variantes en este gen dan lugar al síndrome Kufor-Rakeb con herencia AR, del que se conocen aproximadamente 50 mutaciones. El comienzo de los síntomas suele darse en la adolescencia y se presenta en forma de parkinsonismo, distonía, alteraciones en el movimiento ocular, demencia y trastornos psiquiátricos.
-
Metabolismo del hierro
Los genes CP y FTL desempeñan un papel esencial en la regulación del hierro en el organismo, y variantes en éstos causan formas NBIA en las que su metabolismo está alterado.
CP codifica la ceruloplasmina, una enzima ferroxidasa que funciona como una exportadora de hierro de las células. El Fe2+ intracelular es transportado por la ferroportina hacia la transferrina, vía la actividad ferroxidasa de la ceruloplasmina (Fe2+ →Fe3+). Mutaciones en este gen causan aceruloplasminemia, con herencia AR, aunque pacientes portadores en heterocigosis pueden presentar fenotipos más leves. Alrededor de 70 variantes asociadas a CP han sido descritas. Los síntomas comienzan en edad adulta y comprenden alteraciones cognitivas, ataxia cerebelar, discinesia craneofacial, degeneración retinal e incluso diabetes. Se identifica hipointensidad en estriado, tálamo y dentado.
FTL codifica la cadena ligera de la ferritina, que en humanos es un heteropolímero compuesto por dos subunidades: FTH1 y FTL. La ferritina elimina el hierro ferroso (Fe2+) cuando se encuentra en exceso en las células, de esta manera se evita la formación de ROS, la peroxidación lipídica, la agregación de proteínas y el daño celular. Este hierro es almacenado en forma de óxido férrico (Fe3+) por la ferritina, que lo libera cuando es requerido por las células. Se han descrito diez duplicaciones en el exón 4 de este gen causando neuroferritinopatía, con herencia autosómica dominante (AD), en la que los síntomas aparecen en edad adulta y que comprenden deterioro cognitivo, alteraciones psiquiátricas, ataxia cerebelar, parkinsonismo o blefaroespasmo. Se identifica hipointensidad en los ganglios basales, principalmente en el globo pálido y la sustancia negra.
-
Síndrome Woodhouse-Sakati
El síndrome Woodhouse-Sakati es una enfermedad causada por mutaciones en el gen DCAF17 que presentan una transmisión AR; 17 variantes han sido reportadas hasta la fecha. Este gen codifica una proteína nucleolar cuya función es desconocida, pero pertenece a la familia de genes Dcaf, involucrados en apoptosis, metilación del DNA y regulación del ciclo celular. Las manifestaciones clínicas son complejas, ya que es una enfermedad multisistémica que comprende hipogonadismo, alopecia, diabetes mellitus, sordera, discapacidad intelectual y alteraciones extrapiramidales. En estos pacientes se observa hipointensidad en globo pálido y sustancia negra.
NUEVA FORMA NBIA Y TRASTORNOS RELACIONADOS
En nuestro grupo, hemos identificado una nueva forma parkinsoniana NBIA, causada por mutaciones en el gen FBXO7, y hemos ampliado el cuadro clínico asociado con el gen NR4A2, implicado en un síndrome de distonía-parkinsonismo que solapa con las formas NBIA.
FBXO7 codifica una proteína involucrada en el sistema ubiquitina-proteasoma y en la mitofagia. FBXO7 también es responsable de reclutar la parkina hacia las mitocondrias y comenzar la mitofagia. Mutaciones en este gen se han descrito como una causa rara del síndrome parkinsoniano-piramidal.
En nuestra serie se evaluó una paciente, una mujer de 21 años hija de padres consanguíneos, con un complejo fenotipo que incluía epilepsia, síndrome parkinsoniano-piramidal progresivo, deterioro cognitivo e imágenes MRI que sugerían un patrón NBIA (Correa-Vela et al., 2020). El estudio mediante un panel de diseño propio con 498 genes implicados en trastornos del movimiento (MovDisord-498) reveló que la paciente era portadora de una mutación novel en homocigosis en FBXO7: c.368C>G (p.S123*). Se validó el posible impacto funcional de la variante en fibroblastos de la paciente y se pudo comprobar la ausencia de RNA de FBXO7 y en consecuencia, una alteración en la actividad del proteasoma y una acumulación de proteínas poli-ubiquitinadas. Por tanto, este nuevo fenotipo asociado a FBXO7 es el resultado del sistema de degradación de proteínas alterado, junto al depósito anormal de hierro en el cerebro.
En cuanto a NR4A2, este gen codifica un factor de transcripción nuclear que se expresa principalmente en la sustancia negra. Mutaciones en NR4A2 se han asociado a enfermedades neurodegenerativas, y recientemente de manera más concreta, a un síndrome de distonía-parkinsonismo con discapacidad intelectual de inicio temprano.
En nuestro trabajo (Jesús et al., 2021) se describe un paciente de 30 años, sin antecedentes de enfermedad neurológica en su familia, que sufre distonía-parkinsonismo persistente (DPS), discapacidad intelectual, trastorno del lenguaje y tics motores, de inicio en la infancia. Mediante secuenciación de exoma (Whole Exome Family Plus test; Blueprint Genetics) se identificó la variante novel c.956G>A (p.R319Q) en NR4A2. La variante se clasificó como probablemente patológica siguiendo las recomendaciones del American College of Medical Genetics and Genomics.
Con la descripción de este caso se amplía el espectro fenotípico asociado a NR4A2, así como el tipo de mutaciones implicadas, ya que describimos la primera variante de cambio de aminoácido asociada a un síndrome de distonía-parkinsonismo.
En conclusión, la variabilidad fenotípica entre los pacientes dificulta en gran medida el diagnóstico clínico de casos NBIA, resultando el análisis genético esencial para alcanzar un diagnóstico certero en cada caso. Aun así, la elevada heterogeneidad genética presente dificulta esta tarea, así como el reducido número de casos descritos al tratarse de enfermedades raras, ya que se necesita de la descripción de nuevos casos para establecer una correcta relación genotipo-fenotipo. Todo ello nos hace pensar que existen genes NBIA por descubrir y a la par, hay genes conocidos que no se han asociado a un patrón NBIA en neuroimagen todavía. Un puzle complicado de resolver.


