
INTRODUCCIÓN
Los microbios fueron los primeros organismos en ser secuenciados, debido al pequeño tamaño y la simplicidad de sus genomas. El primer organismo fue el bacteriófago MS2, con un genoma viral de ARN, secuenciado en 1976 (Fiers et al., 1976). Un año después, se secuenció el primer organismo con genoma de ADN, el bacteriófago PhiX174, gracias al trabajo de Fred Sanger y colaboradores (Sanger et al., 1977). Dos décadas más tarde, Craig Venter y su equipo secuenciaron el primer genoma bacteriano, Haemophilus influenza, en 1995, y la primera arquea, Methanococcus jannaschii, en 1996 (Fleischmann et al., 1995; Bult et al., 1996). Ese mismo año, se publicó la secuencia del primer genoma eucariota, Saccharomyces cerevisiae (Johnston, 1996). A pesar de que hace menos de 50 años desde que se secuenció el primer microorganismo, en la actualidad, miles de genomas microbianos se han secuenciado. Este rápido avance ha sido posible gracias al desarrollo y la mejora de las técnicas de secuenciación masiva (next-generation sequencing, NGS). Los primeros métodos de NGS estaban basados en una reacción de amplificación seguida de una secuenciación por síntesis. Hoy en día, también están disponibles en el mercado nuevas metodologías de secuenciación de molécula única. Además, el desarrollo de instrumentos portátiles ha contribuido a disminuir los costes y el tiempo de la secuenciación.
Esta revolución genómica ha transformado el panorama de la microbiología clínica, dando lugar a la nueva especialidad de genómica microbiana. La genómica microbiana es un campo emergente que ha superado a las técnicas tradicionales en la identificación de patógenos, estudio de mecanismos de resistencia y virulencia, rastreo de vías de transmisión, seguimiento de brotes, y monitorización de la aparición y evolución de patógenos emergentes (Figura 1). Este ensayo expone las diversas aplicaciones de la genómica microbiana en la rutina clínica de las enfermedades infecciosas y los servicios de salud pública, así como las limitaciones y barreras que dificultan su total implementación. Cabe destacar que la genómica microbiana está siendo de especial relevancia en la actualidad, siendo clave en el manejo de la crisis ocasionada por la pandemia de la COVID-19.
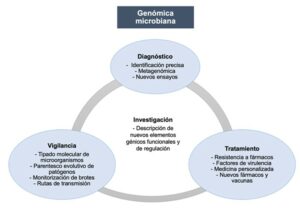
IMPLEMENTACIÓN DE LA GENÓMICA EN LA MICROBIOLOGÍA DIAGNÓSTICA
La identificación específica del patógeno causante de una infección es crucial para el manejo clínico, ya que informa sobre su potencial patogénico. Tradicionalmente, esta identificación se ha basado en características fenotípicas. Las técnicas de tinción, evaluación de la morfología, crecimiento en medios selectivos, test bioquímicos o ensayos inmunológicos constituyen algunas de las técnicas más empleadas en microbiología diagnóstica en los últimos años. Más tarde, se introdujeron los métodos genotípicos. En dicho contexto, para la identificación de virus, se suelen emplear técnicas de PCR (reacción en cadena de la polimerasa) cuantitativa o PCR multiplex (Roy et al., 2017; Yang et al., 2018), como es el caso del SARS-CoV-2 (Corman et al., 2020). Para bacterias y hongos, se lleva a cabo la secuenciación de los genes de ARN ribosomales 16S o 18S, (Wang et al., 2014; Watts et al., 2017). Y recientemente, se ha utilizado también la espectrometría de masas en microbiología diagnóstica, un método rápido y sensible que permite identificar microrganismos por su espectro de proteínas (Singhal et al., 2015).
Hoy en día, gracias al desarrollo de secuenciadores portátiles, el genoma completo de un patógeno se puede obtener suficientemente rápido en el laboratorio, de manera que la secuenciación de genoma completo (whole-genome sequencing, WGS) también se puede utilizar con fines diagnósticos (Didelot et al., 2012). La WGS confiere el nivel más elevado de resolución y discriminación en términos de identificación de patógenos, ya que puede diferenciar entre organismos genéticamente relacionados que no se podrían distinguir por medio de otras técnicas. Como ejemplo de ello, la WGS es el método más preciso para diferenciar entre cepas específicas de Neisseria meningitidis serogrupo B (Jones et al., 2016). También ha sido útil en la detección de infecciones mixtas de Mycobacterium tuberculosis, donde pueden coexistir diferentes genotipos bacterianos (Sobkowiak et al., 2018). Además, la WGS es una técnica universal que se puede aplicar a todo tipo de microorganismos, por lo que un único método sustituiría a todas las técnicas de diagnóstico tradicionales que son específicas para cada especie (Figura 2). Incluso se han desarrollado nuevas metodologías que permiten preparar librerías para la secuenciación de ADN a partir de colonias bacterianas aisladas, evitando la necesidad de realizar un cultivo, lo que constituye un gran beneficio para microorganismos de crecimiento lento o no cultivables (Köser et al., 2014). Además, la WGS de patógenos virales también se puede llevar a cabo directamente a partir de muestras clínicas (Depledge et al., 2011).
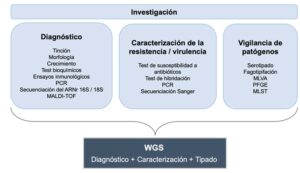
PCR, reacción en cadena de la polimerasa. ARNr, ARN ribosomal. MALTI-TOF, Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight. MLVA, multiple-locus variable number tandem repeat analysis. PFGE, Electroforesis en Gel de Campo Pulsado. MLST, tipificación multilocus de secuencias.
Más allá de la WGS, la metagenómica constituye un método genérico y objetivo que no requiere cultivo previo y que también se ha utilizado con éxito en microbiología diagnóstica. Por ejemplo, la metagenómica es efectiva detectando cualquier virus causante de infecciones del tracto respiratorio a partir de hisopos nasofarígenos (Graf et al., 2016). Un caso de especial relevancia fue un análisis metagenómico de una muestra de fluido cerebroespinal de un paciente de 14 años con una inmunodeficiencia combinada severa. Dicho análisis reveló una infección de Leptospira, lo que condujo a que se tratara al paciente con una terapia antimicrobiana específica (Wilson et al., 2014). Otra contribución relevante de la metagenómica fue el descubrimiento de que cambios en la microbiota intestinal pueden detectar de manera precoz una infección sanguínea de un enterococo resistente a la vancomicina (Ubeda et al., 2010). Además, hoy en día, también se realizan análisis metagenómicos de muestras ambientales que permiten identificar patógenos y que son de gran utilidad en la evaluación del riesgo microbiológico ambiental (Israeli et al., 2019; Yang et al., 2020).
Por otro lado, los datos genómicos se pueden utilizar para desarrollar nuevos ensayos diagnósticos o mejorar los métodos existentes. Por ejemplo, la WGS de Staphylococcus aureus llevó al descubrimiento de un nuevo gen de resistencia homólogo al gen de resistencia a la meticilina mecA, que resultó en la modificación del ensayo de la PCR diagnóstica, de manera que se pudiese detectar esta nueva cepa con el nuevo mecanismo de resistencia (Paterson et al., 2014). Asimismo, la WGS se ha empleado también en la evaluación del rendimiento de los ensayos de diagnóstico molecular (Van Rensburg et al., 2016). Otra de sus aplicaciones clave es el estudio de patógenos en otras especies cercanas al ser humano para tratar de identificar microorganismos con capacidad zoonótica y poder desarrollar estrategias de intervención que permitan controlar potenciales enfermedades infecciosas emergentes (Haagmans, et al, 2009).
Sin embargo, aunque el coste y el tiempo de la WGS y la metagenómica han disminuido considerablemente, actualmente, no es rentable el uso de la genómica para el diagnóstico de todo tipo de infecciones microbianas. Los métodos fenotípicos y genotípicos tradicionales constituyen un procedimiento rápido y económico que son efectivos para la mayoría de los microorganismos comunes. Sin embargo, la genómica se debería implementar para el diagnóstico de patógenos especiales, como microorganismos multirresistentes o microbios complejos que no se puedan identificar de manera precisa por métodos tradicionales y requieran técnicas con un mayor poder discriminatorio.
CONTRIBUCIÓN DE LA GENÓMICA AL TRATAMIENTO DE LAS ENFERMEDADES INFECCIOSAS
Los microorganismos resistentes a fármacos constituyen una amenaza muy seria para la salud pública. La presión evolutiva favorece el desarrollo de mecanismos de resistencia a fármacos, que ocurre principalmente por transferencia genética horizontal y/o la adquisición de mutaciones. Estos mecanismos incluyen la expulsión de los fármacos por medio de bombas de eflujo, evitando su entrada con una barrera impermeable, inactivando o destruyendo el compuesto mediante reacciones enzimáticas o alterando la diana del fármaco. Asimismo, el uso inapropiado de tratamientos antimicrobianos, principalmente antibióticos, está contribuyendo en gran medida a la expansión de los patógenos resistentes (Holmes et al., 2016).
Por ello, una vez se identifica el patógeno responsable de una infección, en el laboratorio, se realizan pruebas para detectar resistencias a fármacos y, de esta manera, guiar la decisión terapéutica, lo que contribuye a maximizar la utilidad de los fármacos actuales. Las pruebas de sensibilidad o antibiogramas se llevan a cabo de manera rutinaria para determinar la susceptibilidad del agente patógeno frente a una serie de medicamentos antimicrobianos, exponiendo dicho microorganismo a una concentración estandarizada del fármaco en cuestión. En la mayoría de los casos, se emplean test fenotípicos rápidos y económicos. Para algunos organismos de crecimiento lento, como M. tuberculosis, los test genotípicos son una alternativa más rápida, aunque se recomienda una confirmación fenotípica. Por ejemplo, el Test de Hain o el Test GenoFlow DR-MTB Array se basan en el uso de sondas de hibridación específicas para mutaciones que causen resistencia (Barnard et al., 2008; Molina-Moya et al., 2016). Para aquellas infecciones víricas para las que se dispone de medicación antiviral, también se recomienda que se realice un test de resistencia a fármacos. En el caso del virus de la inmunodeficiencia humana (VIH), estos test normalmente se basan en la secuenciación de los genes de la proteasa, retrotranscriptasa e integrasa, ya sea por RT-PCR, por secuenciación Sanger o por un procedimiento de NGS guiado (Clutter et al., 2016; Tzou et al., 2018). Por otro lado, la detección de determinantes de virulencia es menos común, pero es importante en algunos casos. Un ejemplo de ello es la detección de la presencia de genes de toxina en las infecciones de Clostridium difficile, que normalmente se lleva a cabo por medio de PCR (Berger et al., 2018).
No obstante, los test fenotípicos no informan sobre el mecanismo de resistencia, y los ensayos genotípicos a pequeña escala únicamente están dirigidos a un subconjunto de características de resistencia o virulencia conocidas. Por ello, la WGS es mucho más útil, ya que no solo predice resistencia a fármacos causada por mecanismos de resistencia conocidos, sino que revela nuevos mecanismos de resistencia y permite caracterizar islas de patogenicidad y elementos genéticos móviles. Asimismo, informa sobre nuevos genes de virulencia, que se pueden detectar comparando datos genómicos con información sobre la manifestación y progresión de la infección, y sobre nuevas funciones génicas y elementos de regulación génica (Figura 2). Toda esta información, normalmente obtenida gracias al papel fundamental del estudio de genomas microbianos en investigación, es de gran utilidad para el manejo clínico, pero también para la salud pública. Además de guiar las decisiones terapéuticas, proporciona nuevo conocimiento y un mejor entendimiento sobre cómo evolucionan los microorganismos y cómo adquieren nuevas características de resistencia o virulencia, que es crucial para el desarrollo de nuevos fármacos antimicrobianos o vacunas.
Por citar algunos ejemplos, la WGS fue de gran éxito en predecir la resistencia a fármacos en M. tuberculosis, mostrando una gran concordancia con test convencionales de susceptibilidad a fármacos y tiempos de entrega más cortos, una vez se implementó en el flujo de trabajo del laboratorio (Iketleng et al., 2018). En las pruebas de resistencia al tratamiento del VIH, la WGS también puede ser un método eficiente para evaluar genotipos resistentes complejos asociados con todos los fármacos antirretrovirales disponibles, y tiene una mayor sensibilidad a la hora de detectar variantes de resistencia de baja frecuencia (Gall et al., 2012). Además, la WGS puede aportar información para el diseño de ensayos clínicos, cambiando los tiempos y/o dosis en casos de adquisición de resistencia mínima, y permiten discernir una reinfección exógena de una recaída de la primera infección (Eyre et al., 2014; Köser, et al., 2014). Asimismo, permite detectar factores de virulencia que son cruciales para entender la patogenicidad microbiana y/o que se pueden emplear como dianas de vacunas, como las cápsulas de polisacáridos en S. aureus. (Ekundayo y Okoh, 2018; Mohamed et al., 2019). Y ciertamente, la gran cantidad de datos de WGS que se han generado del SARS-CoV-2 han sido claves en el desarrollo de las múltiples vacunas de que disponemos en la actualidad (WHO, 2021).
Aparte de la WGS, la secuenciación de ARN y la secuenciación de transposones también se han empleado en investigación para descubrir nuevos determinantes de patogenicidad que se pueden considerar como potenciales dianas terapéuticas, para evaluar mecanismos de resistencia o para estudiar los efectos compensatorios de alteraciones genéticas y el coste de adaptación de la resistencia adquirida (Li et al., 2017; Zhang et al., 2017; van Hensbergen et al., 2018; Xu et al., 2018). Sin embargo, es menos probable que estas técnicas se implementen en la rutina clínica.
Finalmente, aunque existe una correlación alta entre los determinantes genéticos de la resistencia a fármacos y el fenotipo, las pruebas fenotípicas no se deberían remplazar completamente por WGS como técnica rutinaria del laboratorio de microbiología (Bradley et al., 2015; Day et al., 2018). La predicción precisa de resistencia por WGS es complicada debido al conocimiento incompleto de los determinantes genéticos de resistencia y porque todavía quedan mecanismos nuevos por descubrir (Ellington et al., 2017). Sin embargo, la WGS debería implementarse en aquellos casos en los que los mecanismos de resistencia sean importantes para la decisión terapéutica. Por ejemplo, para distinguir los organismos resistentes a los carbapenos debido a carbapenemasas de otros mecanismos de resistencia (Reuter et al., 2013). Además, la WGS sería más rentable que las pruebas fenotípicas de baja escala en el caso de que haya un gran número de marcadores de resistencia que se quiera testar. Y para aquellos patógenos de crecimiento lento, como M. tuberculosis, proporciona resultados de manera más rápida que los test convencionales.
POTENCIAL DE LA SECUENCIACIÓN DE GENOMAS MICROBIANOS PARA LA PREVENCIÓN Y CONTROL DE LAS ENFERMEDADES INFECCIOSAS
Como se ha visto en la actualidad con la pandemia de la COVID-19, en aquellos microorganismos de relevancia epidemiológica, la detección de brotes y el control de la transmisión son cruciales. Tradicionalmente, las técnicas que se han empleado para estudiar el grado de parentesco entre los microorganismos y las rutas de transmisión han sido la PCR, el serotipado, la evaluación de polimorfismos de longitud de fragmentos de restricción o la tipificación multilocus de secuencias (en inglés, MLST) por medio de la secuenciación de un grupo de genes constitutivos o housekeeping. Sin embargo, estas técnicas carecen de suficiente poder de discriminación para distinguir entre microorganismos filogenéticamente muy cercanos.
La genómica aplicada a la epidemiología es el método más completo para identificar relaciones filogenéticas basadas en polimorfismos de nucleótido único (SNP), cosa que es crucial para monitorizar la propagación de patógenos peligrosos a nivel nacional o internacional o para identificar la aparición de nuevas amenazas infecciosas. La combinación de la información genética y geográfica permite trazar la transmisión internacional de patógenos por medio de análisis filogeográficos. En el pasado, estos análisis se hacían de manera retrospectiva, no obstante, la tecnología actual permite monitorizar la transmisión de enfermedades infecciosas a tiempo real. Esto es crucial para el desarrollo de programas de control nacionales e internacionales que implementen vías de intervención tempranas que limiten la diseminación del patógeno y ayuden a cesar la propagación, además de proporcionar información para las estrategias de vacunación. En la actualidad, no cabe duda de que la WGS del SARS-CoV-2 ha sido clave para estudiar la evolución del virus y su epidemiología genética así como para monitorizar su propagación a nivel mundial (Munnink et al., 2020; Pillay et al., 2020; Srivastava et al., 2021). Como ejemplo de ello, proyectos como Nextstrain están siendo cruciales en la monitorización de la evolución del SARS-CoV-2 a nivel mundial, ya que almacenan los resultados procedentes de la WGS de las diferentes variantes mostrando con ello la evolución y propagación del virus (Bedford et al., 2018).
Pero antes de la llegada de la COVID-19, la WGS ya se empleaba en el control de la transmisión de patógenos infecciosos. En particular, la WGS ha revolucionado la detección de brotes causados por patógenos nosocomiales en hospitales, conectando el parentesco entre los patógenos con casos que se solapan en el tiempo y en el espacio (Peacock et al., 2018). Un ejemplo de ello es el uso de datos epidemiológicos y genómicos de S. aureus resistente a la meticilina que revelaron rutas de transmisión que incluían ambientes hospitalarios y comunitarios a escala local, y mostraron cómo este patógeno se propagó desde Reino Unido a Europa y otros continentes (Holden et al., 2013; Coll et al., 2017). Asimismo, los datos genómicos de aislados de Vibrio cholerae en países africanos informaron sobre los diferentes eventos de introducción y las rutas de propagación de este patógeno (Weill et al., 2017). El control del virus del Ébola durante la epidemia en el oeste de África es otro ejemplo de la aplicación de la WGS en brotes a tiempo real. Esto fue posible gracias a la tecnología portátil de secuenciación de Oxford Nanopore, que permite controlar a tiempo real la propagación de infecciones en países en vías de desarrollo (Quick et al., 2016). La WGS también puede revelar el origen de un brote, siendo esto de gran relevancia en infecciones alimentarias (Inns et al., 2015; Butcher et al., 2016). Además, el estudio de la aparición y la propagación de patógenos resistentes a antibióticos tanto temporal como espacialmente, es otra gran contribución, así como medir la evolución y velocidad de crecimiento de la resistencia antimicrobiana (Ford et al., 2013; Safi et al., 2013). Por otro lado, la metagenómica también se puede emplear para identificar patógenos responsables de la aparición de brotes (Loman et al., 2013; Fischer et al., 2014). Además, diferentes patógenos pueden coinfectar a un mismo huésped alterando la fisiopatología de la enfermedad y su transmisibilidad. Se ha visto que las interacciones entre los distintos microorganismos y el orden de infección pueden ayudar a predecir la severidad de potenciales epidemias a partir de modelos multipatogénicos (Clay et al., 2020). En este contexto, la secuenciación de los genomas de dichos patógenos es instrumental para incrementar nuestro entendimiento no sólo de la interacción entre el patógeno y el huésped, sino de las interacciones que ocurren entre los diferentes microorganismos.
En resumen, la genómica no es solo más exacta que las técnicas de tipificación con fines epidemiológicos, sino también más reproducible y estandarizada, ya que un único método sustituiría todos los diferentes, complejos y, a veces, lentos métodos de tipificación, resultando en flujos de trabajo simplificados (Figura 2). Sin embargo, una limitación a tener en cuenta es que una velocidad baja en la tasa de mutación puede impedir el uso de la distancia genética para inferir el parentesco.
Cada país debe determinar la lista de patógenos para la cual se debería realizar WGS con fines de control de transmisión, de manera que su implementación en salud pública sea rentable. Por ejemplo, Salud Pública de Reino Unido emplea WGS para muestras de Escherichia coli, Shigella, Salmonella, Listeria y Campylobacter (Public Health England, 2018). Asimismo, en la actualidad, han creado un consorcio para generar datos de WGS del SARS-CoV-2 que ayude a controlar los brotes en el Reino Unido y proporcione información para guiar la campaña de vacunación (Wellcome Sanger Institute, 2021). Estos datos se pueden almacenar y analizar con fines evolutivos o de control de transmisión. Por ejemplo, se podría emplear machine learning de manera que, en el futuro cercano, los brotes se puedan predecir a través de datos genómicos.
LIMITACIONES DE LA GENÓMICA MICROBIANA Y BARRERAS PARA SU IMPLEMENTACIÓN EN LA RUTINA CLÍNICA Y EN SALUD PÚBLICA
A pesar de que, en la última década, la genómica ha revolucionado la microbiología y las patologías infecciosas, la tecnología actual todavía presenta limitaciones y barreras que limitan su total implementación en la rutina clínica (Tabla 1).
Tabla 1. Principales limitaciones de las técnicas genómicas y principales barreras para su implementación en el ámbito clínico.
|
Limitaciones de la tecnología |
Barreras para la implementación |
|
Coste de la secuenciación |
Análisis bioinformáticos automatizados |
|
Coste del almacenamiento de datos |
Desarrollo de bases de datos de microorganismos |
|
Tiempo de entrega de resultados |
Desarrollo de software especializado |
|
Inversión en infraestructura y equipos |
|
|
Formación |
|
|
Control de calidad de datos |
|
|
Protección de datos |
|
|
Intercambio de datos |
Gracias al desarrollo de secuenciadores portátiles y la consiguiente disminución de los costes de secuenciación y tiempo de entrega de resultados, la genómica no sólo se puede emplear en la investigación sino también en la práctica clínica. Los costes y el tiempo de entrega previstos para la WGS en la rutina clínica se resumen en la Figura 3. No obstante, esta reducción no es suficiente para que se considere asequible la realización de WGS para todo tipo de patógenos, y por ello, cada país debería decidir en qué contexto o para qué microorganismos es rentable aplicar técnicas genómicas. Además, cabe tener en cuenta que una limitación técnica de la secuenciación actual es la incapacidad de cubrir las regiones repetitivas del genoma. Esto puede ser un problema para algunos patógenos, por ejemplo, en el tipado de las regiones repetitivas en tándem de C. difficile (Zaiß et al., 2009). La solución a este problema se basa en la mejora y la implementación de tecnologías de secuenciación de molécula única, como Oxford Nanopore, que permiten una secuenciación completa del genoma de larga lectura.

Otra limitación importante está en el retraso en el desarrollo de la bioinformática, que se encuentra rezagada respecto a los avances en la secuenciación. Los costes relativos al almacenamiento, procesamiento y análisis de datos sobrepasan los costes de secuenciación, y el análisis de datos constituye el cuello de botella en la obtención de los resultados. En este contexto, el desarrollo y la mejora de bases de datos de microorganismos patógenos que contengan genomas de referencia, información sobre factores de resistencia y virulencia, así como la inclusión de datos epidemiológicos y clínicos, permitiría una importante aceleración en el análisis de datos. Concretamente, el desarrollo de software automatizados para análisis de datos genómicos microbianos contribuiría enormemente a la reducción de los costes y los tiempos de entrega.
Otro aspecto que dificulta la implementación de la genómica en microbiología son las cuestiones éticas. Aunque es cuestionable quién tiene la propiedad de los datos genómicos de microorganismos, estos datos pueden informar de manera indirecta sobre contactos personales, por lo que se debe asegurar su protección y anonimización. Además, se debería desarrollar un nuevo consentimiento informado, que no incluya aspectos técnicos sino la información que se puede obtener por medio del uso de técnicas genómicas en enfermedades infecciosas, de manera que los pacientes puedan entender las implicaciones. Otra cuestión de relevancia es el control de calidad, la robustez de los datos y la fiabilidad de los resultados, ya que los datos genómicos tienen implicaciones en el manejo de los pacientes. Además, este tipo de datos normalmente se publica para que puedan ser de utilidad para toda la comunidad científica. Por ello, con el fin de asegurar su calidad y fiabilidad, se requiere una acreditación y validación de todos los pasos en la adquisición y análisis de dichos datos.
A la hora de implementar la genómica en la microbiología clínica se necesita una estrategia nacional. En este sentido, el sistema de Salud Pública de Inglaterra podría servir como modelo (Public Health England, 2018). Para implementar la genómica microbiana en el laboratorio (Figura 4), se requiere una inversión considerable en infraestructura y equipos, y de formación del personal. Por otro lado, se necesita automatización, que actualmente se ha establecido en la extracción de ADN y en la preparación de librerías, pero no en el análisis de datos, por lo que se requiere personal con una alta formación en bioinformática para el desarrollo de pipelines de análisis. Asimismo, cada país debería decidir entre un sistema centralizado basado en laboratorios de referencia, o una estrategia descentralizada basada en centros locales, que sería más costoso, pero implicaría un menor tiempo de entrega de resultados. Por ejemplo, en Reino Unido, la estrategia actual está basada en varios laboratorios de microbiología de referencia, donde se extraen las muestras, y un centro de secuenciación de referencia, cuyos resultados se reportan de vuelta al laboratorio de microbiología para su posterior análisis. Los datos se almacenan por medio de un sistema de manejo de información de laboratorio (LIMS) y se analizan por medio de una infraestructura bioinformática, que junto con los datos de LIMS producen el informe final (Public Health England, 2018).
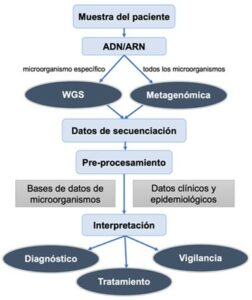
Finalmente, se necesita una colaboración internacional con el objetivo de mejorar la salud pública global. Hay que fomentar el intercambio de datos de manera que exista una mayor información acerca de microorganismos infecciosos y las vías de transmisión. Dicha colaboración puede verse dificultada por aspectos sociales y políticos. No obstante, cabe recordar que la transmisión de patógenos carece de fronteras, por lo que es clave que exista un esfuerzo internacional a la hora de enfrentarnos a epidemias y pandemias.
La relevancia que tiene la colaboración internacional en la lucha frente a agentes infecciosos ha quedado demostrada en la actual pandemia de la COVID-19, en la que dicha colaboración ha sido clave a la hora de controlar la transmisión del virus y en el desarrollo de diferentes vacunas en un periodo de tiempo muy corto. Sin embargo, esta colaboración era escasa antes de la llegada de esta pandemia, por lo que esperemos que sirva de precedente para mantener una colaboración internacional en el control de los microorganismos infecciosos, que nos permita luchar contra otros patógenos actuales comunes, así como con otros microorganismos infecciosos que puedan aparecer en el futuro.
CONCLUSIÓN
La genómica ha transformado la manera en que se abordan las enfermedades infecciosas desde la perspectiva clínica y de salud pública. Los avances en secuenciación han acelerado la transición desde un análisis retrospectivo a prospectivo. Esto ha resultado en un diagnóstico más preciso, un tratamiento más dirigido basado en características de resistencia y/o virulencia, y ha transformado la epidemiología, permitiendo trazar de manera precisa las vías de transmisión a escala local y global.
En la actualidad, no resultaría rentable sustituir todos los test fenotípicos o genotípicos por técnicas genómicas para todos los patógenos de un laboratorio de microbiología. Sin embargo, la implementación de la WGS ha aumentado en el caso de patógenos de interés epidemiológico y la metagenómica se está abriendo su camino en los laboratorios de diagnóstico. El aspecto clave es que la genómica proporciona una información completa con un único test. Por ello, es de gran utilidad en el diagnóstico y caracterización del patógeno y en el control de la salud pública. Desde este punto de vista, se convierte en una técnica rentable y útil.
En el futuro cercano, con la llegada de técnicas de secuenciación y plataformas bioinformáticas más automatizadas, precisas, económicas y rápidas, la genómica se podrá integrar de manera íntegra en los flujos de trabajo de laboratorio y sustituirá a los métodos tradicionales para controles de infecciones rutinarias y manejo de pacientes. Sin embargo, esta transformación requiere un mayor conocimiento y entendimiento de los genomas microbianos y la correlación genotipo-fenotipo.
Finalmente, el objetivo sería, mediante un esfuerzo colaborativo, lograr no solo una salud humana óptima, sino también la salud animal y ambiental, concepto que se conoce como ‘una sola salud’, ya que las tres son interdependientes (FAO, 2021). Estos avances son solo la punta del iceberg. Las contribuciones futuras de la genómica en las enfermedades infecciosas están todavía por descubrir.
Declaración de ausencia de conflictos de intereses
Nada a declarar.
