INTRODUCCIÓN
La edición del genoma tiene el potencial de revolucionar el tratamiento de las enfermedades cardiovasculares crónicas, como, por ejemplo, la enfermedad coronaria. Mientras que los tratamientos farmacológicos existentes hasta el momento requieren la administración de comprimidos diarios o de inyecciones cada pocas semanas o meses (durante el resto de la vida, para poder acumular un beneficio terapéutico completo), la edición del genoma implica cambios permanentes a nivel del ADN y ofrece la posibilidad de tratamientos de una única aplicación, que conferirían una protección duradera contra la enfermedad. Estos tratamientos tendrían la doble ventaja de maximizar el efecto terapéutico (sin dosis perdidas) y eliminar la falta de adherencia a la medicación (un fenómeno extremadamente común). En los últimos años se han obtenido muchos datos prometedores que apuntan a un futuro en el que muchos pacientes se beneficiarán de las terapias de edición del genoma.
Además de resumir la variedad de estrategias de edición del genoma disponibles, esta revisión se centra en el gen para el cual existen más estudios de prueba de concepto, PCSK9 (regulador de los niveles de colesterol en sangre y uno de los motores del riesgo a tener enfermedad coronaria), con el objetivo de ilustrar al lector en las diferentes formas en las que se está avanzando en el desarrollo de terapias de edición del genoma para enfermedades cardiovasculares.
ESTRATEGIAS DE EDICIÓN DEL GENOMA
Cualquier discusión sobre las terapias de edición del genoma debería empezar con una descripción de la variedad de herramientas de edición disponibles en estos momentos. La primera generación de herramientas, conocidas como nucleasas de diseño, tiene dos tipos de funcionalidad: la habilidad de buscar y unirse de forma específica a una secuencia genómica diana, y la capacidad de generar un corte en la doble cadena de ADN en esa secuencia. Las herramientas de edición más recientes han surgido de separar estos dos tipos de funcionalidad y emparejar la capacidad de buscar y unirse al ADN con diferentes actividades que permiten modificar los genes, como son la modificación química de las bases de ADN (edición de bases), la modificación de la expresión del gen (edición del epigenoma) y la transcripción reversa, utilizada para introducir nuevas secuencias de ADN copiadas de un ARN molde (prime editing o edición de calidad). Cada una de estas estrategias se describe brevemente en los siguientes apartados. Una exposición más exhaustiva de las herramientas de edición del genoma, su investigación y sus aplicaciones clínicas, junto a un conjunto de referencias, que la necesidad de brevedad en esta revisión no permite incluir, está disponible en Musunuru, 2021 (ver bibliografía).
Edición con nucleasas
Existen cuatro tipos principales de nucleasas de diseño: las nucleasas con dedos de Zinc (ZFN), las nucleasas efectoras parecidas al activador transcripcional (TALENs), las meganucleasas y los sistemas asociados a repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas CRISPR-Cas. Cada una de ellas tiene la capacidad de buscar secuencias genómicas específicas e introducir cortes de doble cadena en el ADN, aunque los mecanismos por los que llevan a cabo estas tareas son bastante diferentes. Esta revisión está centrada básicamente en los sistemas CRISPR/Cas, ya que son la clase de nucleasas más utilizada y son los que tienen las mejores perspectivas para su aplicación en terapias cardiovasculares en un futuro próximo.
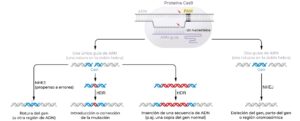
Los sistemas CRISPR/Cas de origen bacteriano que se utilizan ampliamente en la edición del genoma (CRISPR/Cas9 y CRISPR/Cas12) tienen dos componentes, una proteína Cas y un ARN guía (Jinek et al., 2012). El ARN guía proporciona la capacidad del sistema de buscar una secuencia específica y unirse, codificada dentro de la misma secuencia de ARN, mientras que la proteína Cas tiene la capacidad de producir un corte de doble cadena en el ADN, utilizando uno o dos dominios para cortar las dos cadenas de ADN.
La proteína Cas9 de Streptococcus pyogenes o SpCas9, fue el primer sistema CRISPR/Cas en ser adaptado para la edición del genoma en células de mamífero (Figura 1) (Cong et al., 2013; Mali et al., 2013; Cho et al., 2013; Hwang et al., 2013; Jinek et al., 2013). Su ARN guía, de unos 100 nucleótidos de longitud, codifica la especificidad para el ADN diana en los primeros 20 nucleótidos, en una zona denominada espaciador. SpCas9 se une a los otros 80 nucleótidos del ARN guía y, formando un complejo proteína-ARN, escanea cualquier molécula de ADN con la que contacta. Durante este proceso de escaneo SpCas9 se detiene en los motivos NGG del ADN (siendo N cualquier nucleótido) y posiciona el espaciador del ARN guía frente a la cadena de ADN que no contiene el motivo NGG, llamada cadena diana. Si hay una complementariedad perfecta (o en algunos casos, casi perfecta) entre la secuencia de la cadena diana y la secuencia del espaciador, se produce un apareamiento de bases de Watson-Crick extensivo entre el ADN y el ARN que activa a SpCas9 y resulta en un corte de la doble cadena proximal al tercer par de bases aguas arriba del motivo NGG. La secuencia de ADN de la cadena no diana correspondiente a la secuencia del ARN espaciador se denomina el protoespaciador, y está constituida por los 20 nucleótidos aguas arriba del motivo NGG. El motivo NGG se denomina motivo adyacente al protoespaciador (PAM).
SpCas9 se ha convertido en la herramienta de edición del genoma más popular debido tanto a la facilidad con la que se puede redireccionar a cualquier secuencia genómica deseada, simplemente cambiando las 20 bases del espaciador del ARN guía, como a sus elevadas tasas de rendimiento de edición, comparada con otras herramientas de edición genómica (Ding et al., 2013). No obstante, también se utilizan proteínas Cas9 adaptadas de otras especies bacterianas para la edición del genoma, principalmente la proteína Cas9 de Staphylococcus aureus (SaCas9), que tiene la doble ventaja de ser más pequeña que SpCas9 y de tener una secuencia PAM diferente (NNGRRT, donde R puede ser G o A), lo que le da un rango de diana diferente (Ran et al., 2015). La modificación proteica de SpCas9 y SaCas9 ha producido variantes que reconocen nuevas secuencias PAM (Kleinstiver et al., 2015a, 2015b). Al menos otras tres proteínas Cas, todas de la familia Cas12, han demostrado ser editores del genoma eficaces: Cas12a/Cpf1, Cas12b/C2c1, y Cas12e/CasX (Zetsche et al., 2015; Strecker et al., 2019; Liu et al., 2019).
El resultado final de la edición mediante nucleasas depende de la forma en la que cada célula repara el corte de doble cadena introducido por la nucleasa. El mecanismo de reparación por defecto es la unión de extremos no homólogos (NHEJ), en la que los extremos de ADN libres se unen entre sí (Figura 1) (Rouet et al., 1994; Bibikova et al., 2002). Aunque a menudo se restaura la secuencia original de ADN, el NHEJ es un proceso propenso a errores, que puede introducir inserciones o deleciones (mutaciones indel), normalmente de uno o unos pocos pares de bases, pero a veces, de docenas, cientos o incluso miles de pares de bases. Debido a que las mutaciones indel se producen de una manera semialeatoria, diferentes células adquirirán distintos indels. A pesar de esta impredecibilidad en la mutagénesis, si el objetivo es simplemente interrumpir un gen o un elemento no codificante, estrategia adecuada para diversos trastornos cardiovasculares como la enfermedad vascular aterosclerótica o la amiloidosis por trastiretina, el NHEJ es muy apropiado, llegando hasta un 100% de eficacia de edición en algunos contextos. Si el objetivo es hacer un cambio preciso a través de la edición por nucleasas, como la corrección de una mutación causante de una enfermedad, se debe utilizar un mecanismo de reparación celular diferente, la reparación dirigida por homología (HDR) (Figura 1) (Rouet et al., 1994; Bibikova et al., 2002, 2003). La HDR tiene varias limitaciones importantes. Por una parte, requiere un molde extra para la reparación del ADN, además de la proteína Cas y del ARN guía, lo que complica su introducción en las células. En segundo lugar, su rendimiento es típicamente menor que el conseguido mediante NHEJ, en muchos casos con un porcentaje muy bajo. Y además es menos activa en las células no proliferativas que en las células proliferativas, lo que hace que sea menos práctica para ser utilizada en órganos implicados en enfermedades cardiovasculares, como el corazón o el hígado. Afortunadamente, las últimas estrategias de edición del genoma como la edición de bases o prime editing ofrecen la precisión de la HDR superando sus limitaciones, como se describe más adelante.
La edición mediante nucleasas en el contexto de las aplicaciones terapéuticas puede tener dos tipos de consecuencias no deseadas: la introducción de cambios no deseados en el sitio diana, como mutaciones indel grandes e incluso anomalías cromosómicas, y la edición fuera de la diana (denominada off-target), debido a que las nucleasas se pueden unir a regiones que no coinciden perfectamente con el espaciador del ARN guía, lo que resulta en mutaciones indel en posiciones distintas a las que se pretendía. En teoría si se produjera una mutación de este tipo en genes supresores de tumores o en oncogenes podría aumentar el riesgo de cáncer a largo plazo. La edición en posiciones no deseadas se puede reducir mediante alteraciones en la proteína Cas o en el ARN guía, pero muchas veces esto también reduce la edición en la diana objetivo (Kleinstiver et al., 2016; Slaymaker et al., 2016; Fu et al., 2014).
Edición de bases
La edición de bases, al igual que la edición del epigenoma y la edición de calidad, aprovecha el hecho de que CRISPR/Cas9 se puede dirigir a una posición específica del genoma aunque los dominios de corte de Cas9 hayan sido alterados de manera que solo puedan cortar una (nickase Cas9 o nCas9) o ninguna cadena de ADN (Cas9 “muerta” o dCas9). La fusión de dominios adicionales a la nCas9 o a la dCas9 puede añadir diferentes tipos de funcionalidad al sistema CRISPR/Cas9.
Hay dos clases principales de editores de bases: editores de bases de citosina (C), que llevan a que una C de la cadena de ADN sea reemplazada por otra base (normalmente una timina o T, y a veces una guanina o G), (Komor et al., 2016; Koblan et al., 2021) y los editores de bases de adenina (A), que hacen que una A sea reemplazada por una G (Gaudelli et al., 2017) (Figura 2). Si la nCas9 se fusiona con algún dominio citidina desaminasa natural (como por ejemplo el de la proteína APOBEC1 o el de la proteína AID), la desaminasa puede actuar potencialmente sobre cualquier C dentro de la ventana de edición en la cadena de ADN no diana. Esta ventana de edición, cuya extensión varía según el ortólogo de Cas9 utilizado, se define de la siguiente forma: la relajación de las cadenas de ADN por parte de la proteína Cas9 y la hibridación de la cadena diana con el ARN guía crean una estructura llamada R-loop, que lleva a que una parte de la cadena no diana no hibridada forme una burbuja de cadena simple accesible a la acción del dominio desaminasa. La desaminación convierte la C en U (uracilo), que en condiciones normales volvería a convertirse en C mediante la acción de la uracilo-ADN glicosilasa, pero la fusión del dominio inhibidor de la uracilo-ADN glicosilasa a la nCas9 evita esta reparación. Cuando la proteína nCas9 hace un corte simple en la cadena diana, la reparación implica la eliminación de nucleótidos alrededor del sitio de corte, seguida por el reemplazo de los nucleótidos mediante complementariedad con la cadena no diana. Por cada U presente en la cadena no diana se coloca una A en la posición complementaria en la cadena diana (debido a que una A y una U pueden formar un par de bases). Después de reparar el corte, la célula reemplaza la U no estándar (que normalmente solo se encuentra en el ARN) por la T estándar. De esta manera, un par de bases C-G cambia a un par de bases T-A.
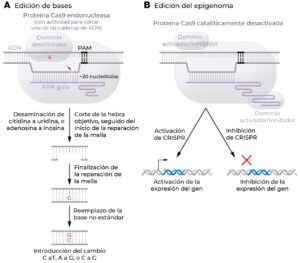
Debido a que no existe una adenosina desaminasa de origen natural que actúe sobre cadenas simples de ADN, se ha utilizado la evolución de proteínas en el laboratorio para crear una nueva ADN-desaminasa (Gaudelli et al., 2017). La fusión entre este dominio de desaminasa evolucionado y la nCas9 permite la edición de bases de la adenina, que opera de forma similar a la edición de bases de la citosina. Dentro de la ventana de edición de la cadena no diana, la A se convierte en I (inosina). A continuación, se repara el corte en la cadena diana y se coloca una C en la cadena diana opuesta a la I, y la I no estándar es reemplazada por una G estándar. De esta manera, un par de bases A-T cambia a un par de bases G-C.
Los editores de bases de citosina y adenina están limitados por los tipos de ediciones que pueden producir: cambios de un único nucleótido, mayoritariamente transiciones. Pero si la edición deseada (por ejemplo, la introducción de una mutación sin sentido para interrumpir un gen, o la corrección de una mutación causante de una enfermedad) es compatible con la edición de bases, se puede conseguir una alta eficacia, en algunos casos casi del 100%, incluso en células que no están en división. La edición en posiciones no diana (off-target) puede ocurrir, pero normalmente está limitada a cambios en un único nucleótido que resultan de la actividad desaminasa.
Edición del epigenoma
Si la proteína dCas9 se dirige a la secuencia del promotor de un gen o a una región intensificadora de la transcripción (enhancer), tiene el potencial de interferir a nivel estérico con los factores que normalmente interactúan con esa secuencia (Qi et al., 2013). Este fenómeno llamado interferencia CRISPR – por analogía con la interferencia de ARN mediada por ARN de horquilla cortos (short hairpin RNAs), aunque el mecanismo es bastante diferente – se puede explotar para bloquear la expresión de genes específicos. La interferencia CRISPR es más potente si dCas9 está fusionada con un dominio, como el KRAB (Krüpel-associated box), que reprime activamente la expresión del gen a través de la modificación de la estructura local de la cromatina (Figura 2) (Gilbert et al., 2013). El fenómeno opuesto, llamado activación CRISPR, se consigue fusionando dCas9 con dominios que potencian la expresión del gen (como el activador transcripcional VP16) o extendiendo la secuencia del ARN guía en su extremo 3′ con aptámeros de ARN capaces de reclutar dominios de activación (Figura 2) (Gilbert et al., 2014). Ni la interferencia CRISPR ni la activación CRISPR producen cambios en la secuencia de ADN y el efecto en la expresión génica parece durar solo mientras está presente la proteína de edición, es decir, el efecto es transitorio.
Un tipo diferente de edición del epigenoma implica la alteración del estado de metilación de las secuencias de ADN, específicamente de las citosinas en las secuencias de dinucleótidos CpG. La metilación cerca del sitio de inicio de transcripción está típicamente vinculada al silenciamiento del gen, mientras que la no metilación está vinculada con la activación génica. Las fusiones de dCas9 con los dominios metiltransferasa o desmetilasa pueden disminuir o aumentar, respectivamente, la expresión del gen. Estos cambios en la metilación pueden mantenerse a largo plazo además de ser reversibles si posteriormente se utiliza en la misma posición genómica un editor epigenómico con el efecto opuesto (Nuñez et al., 2021).
Otros tipos de edición
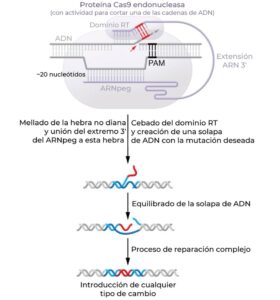
La edición de calidad o prime editing ha sido desarrollada recientemente con el objetivo de superar las limitaciones en los tipos de cambios que se pueden llevar a cabo mediante la edición de bases, así como las limitaciones de la HDR (Anzalone et al., 2019). En la edición de calidad nCas9 se fusiona con una transcriptasa reversa que puede producir una cadena de ADN complementaria a un ARN sustrato de cadena simple. Este sustrato se proporciona como una extensión del extremo 3′ del ARN guía, con una secuencia de ARN complementaria a la cadena de ADN no diana que también incluye una mutación deseada. Este ARN guía extendido se denomina pegARN. La proteína nCas9 corta la cadena no diana y el extremo 3′ del pegARN hibrida con la cadena no diana en un lado del corte (dirección 5′), de manera que se forma un dúplex de ARN-ADN que sirve de molde a la transcriptasa reversa. Esta enzima produce una secuencia de ADN con la mutación deseada en la parte media del pegARN (Figura 3). Esta nueva cadena de ADN puede reemplazar parte de la cadena no diana, lo que resulta en una incorporación permanente de la mutación. Aunque la eficacia de la edición de calidad es más baja que la de la edición mediante nucleasas y NHEJ, o que la de la edición de bases (pero más alta que la de la edición mediante HDR), es capaz de introducir de forma precisa una gran variedad de mutaciones: cambios en un único nucleótido de todo tipo y mutaciones indel de muchas longitudes, hasta docenas de pares de bases y potencialmente incluso más. La extensión de la edición fuera de la secuencia diana (off-target) con los editores de calidad todavía se tiene que definir.
La edición de ARN es una técnica de edición ortogonal en la que se utilizan las proteínas de la familia Cas13 (Abudayyeh et al., 2017). El sistema CRISPR/Cas13, al igual que los sistemas CRISPR/Cas9 y CRISPR/Cas12, tiene componentes proteicos y de ARN, pero actúan sobre dianas de ARN en vez de sobre ADN. Los editores de ARN se pueden utilizar para degradar ARN diana (Abudayyeh et al., 2017) o para editar bases (A por I o C por U) en ARN diana (Cox et al., 2017; Abudayyeh et al., 2019). Debido a la corta vida de las moléculas de ARN, la persistencia de los efectos en el ARN depende de la presencia prolongada del editor, para que pueda continuar actuando sobre las nuevas moléculas de ARN transcritas.
EDICIÓN TERAPÉUTICA DEL GENOMA: EL EJEMPLO DE PCSK9
Casi todas las aplicaciones terapéuticas de la edición del genoma para enfermedades cardiovasculares que se están explorando de forma activa tienen lugar dentro del cuerpo del paciente, en órganos como el corazón o el hígado – aplicaciones in vivo –, en lugar de utilizarse en células extraídas del cuerpo del paciente, que se modifican fuera de él y se vuelven a trasplantar en el paciente – aplicaciones ex vivo. Muchos de los estudios de prueba de concepto para la edición terapéutica del genoma in vivo se han centrado en genes estrechamente asociados a un factor de riesgo causal, modificable y bien establecido, para la enfermedad cardiovascular aterosclerótica, concretamente, el colesterol de lipoproteínas de baja densidad (LDL-C). Entre ellos hay un gen que destaca particularmente: el de la proteína convertasa subtilisina/kexina tipo 9 (PCSK9). Este gen se expresa preferentemente en los hepatocitos del hígado y su producto proteico se secreta al torrente sanguíneo, donde ejerce su actividad primaria de aumentar la concentración de LDL-C en sangre, por lo que se ha convertido en un popular gen diana para probar las nuevas tecnologías de edición del genoma. El hígado es un órgano fácil de alcanzar a través de varios métodos de administración y tanto los niveles de proteína PCSK9 como los niveles de LDL-C en sangre son marcadores farmacodinámicos fáciles de medir, que permiten comprobar la edición del gen PCSK9. Por lo tanto, concentrar la discusión de esta revisión en los
estudios de edición del genoma de PCSK9 proporciona una visión completa del progreso en el campo de la edición del genoma terapéutica e ilustra las decisiones clave que deben tomarse en el diseño de terapias de edición del genoma. No hay otros genes (implicados en enfermedades cardiovasculares u otras enfermedades) para los que exista una cantidad tan grande de datos en estos momentos.
Por sus propios méritos como diana de fármacos, PCSK9 es, por supuesto, un gen de gran interés para el desarrollo de terapias cardiovasculares. PCSK9 se identificó como una causa de la hipercolesterolemia familiar (niveles elevados de HDL-C en sangre de origen genético) gracias al descubrimiento de que pacientes con esta enfermedad tenían una mutación de ganancia de función en una de las copias del gen (Abifadel et al., 2003). Por otra parte, las personas con una mutación sin sentido de pérdida de función en una de las copias del gen tienen niveles sustancialmente reducidos de LDL-C en sangre y hasta un 88% de reducción en el riesgo de enfermedad coronaria (Cohen et al., 2006), sin consecuencias graves para la salud (Rao et al., 2018). Además, también se han descrito algunos individuos con silenciamiento completo de PCSK9 debido a dos mutaciones de pérdida de función (Zhao et al., 2006; Hooper et al., 2007). Estas observaciones han convertido a PCSK9 en una diana de fármacos atractiva para el tratamiento y la prevención de la enfermedad coronaria. Actualmente existen ya tres fármacos dirigidos a PCSK9 aprobados para su uso en pacientes: alirocumab y evolocumab (anticuerpos monoclonales que se unen a la proteína PCSK9 en la sangre), e inclisiran (un pequeño ARN de interferencia que reduce los niveles de ARNm de PCSK9 en los hepatocitos), y hay varios más en proyecto de desarrollo de medicamentos.
En una primera demostración de la alta eficacia de la edición del genoma de mamíferos in vivo, se utilizó un vector adenoviral que contenía un ADN que codificaba para SpCas9 y un ARN guía que tenía como diana una secuencia del exón 1 del gen Pcsk9 de ratón, para inactivar el gen Pcsk9 en hígado de ratón, introduciendo mutaciones de pérdida de función vía NHEJ (Ding et al., 2014). En general, se evita el uso de vectores adenovirales en pacientes, debido al riesgo de que se produzcan respuestas inmunitarias graves y potencialmente mortales. El uso de vectores virales adenoasociados (AAV) es preferible, ya que se toleran mejor inmunológicamente y por tanto son más apropiados para la clínica. Un inconveniente de los vectores AAV es la limitación en su capacidad de carga (<5 kb de secuencia exógena), ya que la mayoría de las herramientas de edición del genoma no caben dentro de ellos. Por ejemplo, la secuencia codificante de SpCas9 tiene unas 4.2 kb y el casete de expresión del ARN guía mide unos 500 pares de bases, lo que deja un espacio restante mínimo para la secuencia del promotor de SpCas9 y la secuencia de poliadenilación. Los vectores adenovirales tienen una capacidad de carga mucho más grande, por lo que se eligieron para este estudio inicial de prueba de concepto de la edición genómica in vivo mediante CRISPR/Cas9. Los investigadores administraron el vector SpCas9 o un vector control a ratones normales. Después de varios días había más del 50% de edición en el sitio diana de PCSK9 en todo el hígado. Los cambios más comunes fueron deleciones o inserciones de uno o dos pares de bases, mientras que los cambios de docenas de pares de bases se dieron con mucha menos frecuencia. La edición de PCSK9 fue acompañada de una disminución del 90% en los niveles de la proteína PCSK9 en sangre y de un 35-40% en los niveles de colesterol en sangre, casi tanto como la reducción del 36-52% del colesterol en sangre observada en ratones mutantes (knockout) para Pcsk9 en la línea germinal (Rashid et al., 2005). En este estudio inicial no se encontró ningún tipo de edición fuera de la diana en una serie de posiciones candidatas no deseadas.
En un estudio posterior, otro grupo de investigadores replicó los mismos resultados de edición en ratones tratados con un vector adenoviral parecido, con SpCas9 y el mismo ARN guía para Pcsk9 (Akcakaya et al., 2018). Los investigadores evaluaron rigurosamente la edición en regiones no deseadas en el hígado mediante una estrategia de dos pasos. En el primer paso se hizo un rastreo de posiciones del genoma candidatas a sufrir edición no deseada (off-target) utilizando una técnica bioquímica llamada CIRCLE-Seq, en la que fragmentos de ADN genómico circularizado de ratón se mezclan in vitro con la proteína SpCas9 y el ARN guía de Pcsk9 y posteriormente se realiza secuenciación masiva paralela para identificar los fragmentos de ADN linealizados. Mediante este procedimiento se identificaron 182 posiciones candidatas. En el segundo paso, los investigadores realizaron una amplificación por PCR de las posiciones candidatas y una secuenciación masiva paralela profunda de los amplicones de las muestras de ADN genómico del hígado de los ratones tratados con SpCas9. No se encontró edición en ninguna de las posiciones, lo que sugiere que es posible seleccionar los reactivos de la edición del genoma con suficiente especificidad de edición para que no se detecte mutagénesis no deseada in vivo.
A pesar de sus resultados esperanzadores, estos estudios no tienen una relevancia directa con lo que podría pasar en la edición de PCSK9 en pacientes humanos, ya que existen tres grandes diferencias entre los ratones y los humanos. En primer lugar, hay diferencias sustanciales entre las secuencias del gen Pcsk9 de ratón y el gen PCSK9 humano, por lo que no se puede identificar una secuencia efectiva de ARN guía que funcione en ambas especies. En segundo lugar, existen diferencias fundamentales entre el genoma humano y el de ratón, de tal manera que el perfil de sitios de edición off-target en el ratón no es predictivo para la edición en posiciones no deseadas en el genoma humano. En tercer lugar, hay diferencias fisiológicas importantes entre los hepatocitos humanos y los de ratón, de forma que los resultados de la edición podrían ser significativamente distintos en los dos tipos celulares.
Para una mejor evaluación de la eficacia y seguridad de una potencial terapia de edición de PCSK9 humano que pueden obtenerse en ratones no modificados, se llevó a cabo un estudio en ratones quiméricos con hígados humanizados, un sistema modelo en el que los hepatocitos endógenos de ratón se reemplazan con hepatocitos humanos trasplantados (Wang et al., 2016). Los ratones con hígado humanizado se trataron con un vector adenoviral que codificaba para SpCas9 y un ARN guía que tenía como diana una secuencia del exón 1 del gen PCSKA9 humano. En este caso, se obtuvo alrededor de un 50% de edición, mediada por NHEJ, de los alelos de PCSK9 humanos presentes en el hígado humanizado, sin edición en varias posiciones off-target candidatas y una reducción de alrededor del 50% en los niveles de proteína PCSK9 humana en sangre.
Los siguientes estudios se alejaron de los vectores adenovirales y se enfocaron en métodos de administración más adecuados para el uso clínico. En el primer estudio en utilizar AAV para obtener una alta eficacia en la edición del genoma de mamíferos in vivo se introdujo, en un único vector AAV, la secuencia de SaCas9 junto con la de un ARN guía, que podía ser cualquiera de los dos ARN guía disponibles que tenían como diana secuencias del gen Pcsk9 de ratón (Ran et al., 2015). Después de la administración de los dos tipos de vectores AAV a ratones normales, se obtuvo un 40-50% de edición de Pcsk9 mediada por NHEJ en todo el hígado, con una reducción de más del 90% en los niveles de proteína PCSK9 en sangre y del 40% de los niveles de colesterol en sangre, resultado muy similar a los efectos observados en los anteriores experimentos, en los que se utilizaron adenovirus para la administración de SpCas9.
Después del éxito del uso de los vectores AAV, se utilizaron métodos no virales para la administración de CRISPR/Cas9 in vivo en el hígado. En el primer estudio de administración de CRISPR/Cas9 en el hígado exclusivamente mediante un vector no viral, se inyectaron de forma seriada, en ratones normales, dos tipos de nanopartículas lipídicas (LNP por sus siglas en inglés) formuladas con el ARNm de SpCas9 o con un ARN guía sintetizado que tenía como diana una secuencia de Pcsk9. Como resultado, se obtuvieron reducciones del 40-50% en los niveles de proteína PCSK9 (Jiang et al., 2017). En un estudio posterior se administró a los ratones normales LNP formuladas con el ARNm de SpCas9 o con una mezcla de dos ARN guías sintetizados que tenían como diana secuencias distintas de Pcsk9 que incluían modificaciones químicas para aumentar la estabilidad del ARN in vivo (Yin et al., 2017). El tratamiento con LNP resultó en más del 80% de edición del gen en todo el hígado. Esta alta tasa de edición es debida a una deleción muy eficaz mediada por NHEJ entre las posiciones diana de los dos ARN guías (Figura 1). El resultado de la edición fue una ausencia de proteína PCSK9 detectable en sangre y una reducción del 35-50% de los niveles de colesterol. No se encontró edición en los pulmones o en el bazo, lo que implica que las LNP se dirigieron específicamente hacia el hígado, o que el locus Pcsk9 solo estaba accesible para la acción de la SpCas9 en las células hepáticas.
En el siguiente conjunto de estudios se exploró el desarrollo de nuevos tipos de edición del genoma, como la edición de bases, la edición epigenómica y la utilización del ARN como diana. Los editores de bases de citosina pueden introducir directamente mutaciones sin sentido en los genes mediante cambios C por T o G por A (este último resulta de las ediciones C por T en la cadena antisentido) en codones específicos. En uno de los trabajos se administró a ratones normales un vector adenoviral que contenía el editor de base de citosinas BE3 junto con un ARN que tenía como diana el triptófano del codón 159 de Pcsk9 (TGG, en el que la edición de una o las dos G por A resulta en un codón de parada) (Chadwick et al., 2017). Como resultado del tratamiento un 30% de los alelos de Pcsk9 del hígado fueron editados, la mayoría hacia el deseado codón de parada, aunque algunas ediciones resultaron en mutaciones no sinónimas, así como mutaciones indel en una tasa del 1-2%. La correspondiente reducción de los niveles de proteína PCSK9 en sangre llegó hasta el 60% y la reducción de los niveles de colesterol fue de alrededor del 30%.
La misma estrategia basada en la edición de bases de Pcsk9 se utilizó en una de las primeras demostraciones de la edición del genoma fetal en ratones. Por analogía con la cirugía fetal, en la que los pacientes con defectos anatómicos potencialmente mortales son tratados cuando todavía se encuentran en el útero materno, la edición genómica fetal estaría reservada para los pacientes con trastornos genéticos graves que causan daños en el estadio prenatal y que resultan en una elevada morbilidad y mortalidad después del nacimiento. En la demostración se administró un vector adenoviral con el editor de bases BE3 que tenía como diana el gen Pcsk9, en el hígado de fetos de ratón mediante una inyección en la vena vitelina, precursora de la vena porta (Rossidis et al., 2018). Este procedimiento prenatal tuvo como resultado una reducción permanente de los niveles de proteína PCSK9 y colesterol en sangre, después del nacimiento. (Hay que tener en cuenta que la hipercolesterolemia no es una enfermedad que requiera, bajo ninguna circunstancia, un tratamiento prenatal y este estudio con ratones se llevó a cabo únicamente como prueba de concepto de la edición del genoma fetal. En el mismo estudio se realizó una edición de bases del gen Hpd, que resultó en un tratamiento exitoso para la tirosinemia hereditaria de tipo 1 en fetos de ratón).
En una demostración de la edición del epigenoma, se fusionó la SaCas9 catalíticamente inactiva con el dominio represor KRAB y tanto el editor como un ARN guía que tenía como diana el promotor de Pcsk9 se introdujeron en dos vectores AAV diferentes (Thakore et al., 2018). En el estudio, los dos tipos de vectores AAV se administraron a la vez a ratones normales, lo que resultó en una reducción del 50% de la expresión hepática de Pcsk9 y en una reducción de alrededor del 80% en los niveles sanguíneos de proteína PCSK9, además de la correspondiente reducción de los niveles de LDL-C en sangre. Cabe destacar que los efectos terapéuticos se fueron debilitando en el transcurso de unos pocos meses, lo que sugiere que conforme disminuyó la expresión del editor del epigenoma también lo hizo la represión de Pcsk9. Otro estudio demostró la utilización de un editor de ARN, CasRx (Cas13d), administrado a través de un vector AAV, para interrumpir la expresión de Pcsk9 (He et al., 2020). En este caso, como en la edición del epigenoma, se esperaría que el efecto terapéutico se prolongase mientras persistiera la expresión del editor. Tanto la edición del epigenoma como la utilización del ARN como diana probablemente requerirían administraciones repetidas del tratamiento para mantener efectos terapéuticos crónicos, a diferencia del efecto de “una única administración necesaria” que se puede obtener con la edición mediante nucleasas o con la edición de bases.
Los trabajos más recientes se han centrado en un paso clave en la transición de la edición terapéutica del genoma a los pacientes humanos: la demostración de la eficacia y seguridad en primates no humanos. En el primer estudio se utilizaron meganucleasas en vez del editor CRISPR para editar el gen PCSK9 (Wang et al., 2018; Wang et al., 2021). Los investigadores utilizaron un vector AAV que codificaba para una meganucleasa específica para una secuencia del exón 7 de PCSK9, que se expresaba mediante un promotor fuerte específico de hígado. Este vector se administró a macacos Rhesus por vía intravenosa en diferentes dosis. En la dosis más alta se observó una edición de PCSK9 del 46% en todo el hígado, con las correspondientes reducciones en sangre del 85% en los niveles de proteína PCSK9 y del 56% en los niveles de colesterol LDL. Las dosis más bajas de AAV produjeron tasas de edición substancialmente más bajas. Estas reducciones persistieron durante al menos 3 años (Wang et al., 2021).
A pesar del éxito del estudio con meganucleasas en primates no humanos, se deben tener en cuenta varios aspectos importantes para la transición a la clínica. En primer lugar, se produjo una edición no intencionada de la diana. Aunque el objetivo era interrumpir el gen PCSK9 mediante NHEJ, lo que resultaría en pequeñas mutaciones indel, el evento de edición más frecuente fue la integración de las secuencias del vector AAV en el genoma, en los sitios de corte de doble cadena en el gen PCSK9, con consecuencias poco claras sobre la seguridad. En segundo lugar, el tratamiento con meganucleasas causó mutagénesis relevante en muchas posiciones del genoma no deseadas, tanto en hígados de monos in vivo como en hepatocitos humanos in vitro. En tercer lugar, se observó una sustancial respuesta inmunitaria de células T, tanto contra el vector AAV como contra la meganucleasa, que causó un aumento moderado de los niveles de transaminasa en sangre en todos los monos tratados, varias semanas después del tratamiento, lo que concuerda con una muerte de hepatocitos inmunomediada. Este aumento de transaminasa se resolvió de forma espontánea tras varias semanas o meses, sin consecuencias aparentes a largo plazo y sin que se atenuara la edición de PCSK9 o la reducción de los niveles sanguíneos de PCSK9 o LDL-C. A pesar de estas cuestiones, este estudio estableció la posibilidad de la edición del genoma a través de terapias de un solo paso en primates, con efectos terapéuticos que perduran varios años y probablemente toda la vida, en los animales tratados.
En dos estudios recientes con primates no humanos, dos equipos de investigadores utilizaron la edición de bases de adenina para interrumpir el gen PCSK9 en macacos cynomolgus (Rothgangl et al., 2021; Musunuru et al., 2021). Para la administración del editor en el hígado se utilizaron LNP que contenían el ARNm de un editor de bases de adenina y un ARN guía sintético que tenía como diana un sitio donador de splicing que se encuentra al final del exón 1 del gen PCSK9. En uno de los estudios se observaron efectos relativamente modestos un mes después del tratamiento: 26% de edición de PCSK9 en todo el hígado, 32% de reducción en los niveles de PCSK9 en sangre y una reducción del 14% en los niveles sanguíneos de LDL-C. En el otro estudio se produjeron efectos farmacodinámicos más elevados e importantes para la transición a la clínica, que duraron más de 8 meses: el 66% de edición de PCSK9 en todo el hígado, un 90% de reducción en los niveles de PCSK9 en sangre y una reducción alrededor del 60% en los niveles de sanguíneos de LDL-C (Musunuru et al., 2021).
Estos estudios contrastan con el estudio realizado con meganucleasas en primates no humanos en diversos aspectos. Puesto que con la estrategia con LNP no se utiliza ningún componente de ADN, no hubo riesgo de integración de la secuencia del vector en el genoma, y la utilización de los editores de bases resultó en un cambio específico de un par de bases en PCSK9 en contraposición a los indels semialeatorios y las inserciones de las secuencias del vector AAV inducidas por la meganucleasa. Con la edición de bases no se encontró ninguna edición genómica no deseada en ninguna de las numerosas posiciones candidatas en los hepatocitos humanos y en el hígado de mono solo se observó un bajo nivel de edición no deseada en una posición candidata, siendo los cambios de una sola base, en lugar de indels. Finalmente, el tratamiento con LNP resultó en un aumento inmediato y transitorio en los niveles de transaminasa en sangre, que se resolvió espontáneamente en una o dos semanas, sin que se diera una transaminitisindicadora de una respuesta inmunitaria robusta, aunque cabe destacar que la administración repetida de LNP en uno de los estudios provocó el desarrollo de anticuerpos contra el editor de bases en algunos animales.
PERSPECTIVAS PARA LA TRASLACIÓN A LA CLÍNICA EN UN FUTURO PRÓXIMO
Tomando como base todos los estudios mencionados anteriormente, parece que la edición de PCSK9 está lista para ser probada en ensayos clínicos con pacientes de hipercolesterolemia y enfermedad coronaria. Y, por supuesto, el potencial terapéutico de la edición genómica para las enfermedades cardiovasculares y otros tipos de enfermedades va más allá de PCSK9 (Tabla 1). Los estudios preclínicos han establecido las perspectivas para el tratamiento de la hipercolesterolemia familiar homocigota mediante la edición del gen ANGPTL3 en hígado (Chadwick et al., 2018). Además, aunque no son enfermedades exclusivamente cardiovasculares, la mortalidad acelerada de los pacientes con distrofia muscular de Duchenne o progeria de Hutchinson-Gilford se debe a complicaciones cardiovasculares que se originan en el corazón o en los vasos sanguíneos y los estudios preclínicos han demostrado una mejora de los fenotipos cardiovasculares mediante la edición con nucleasa Cas9 y la edición de bases (Amoasii et al., 2018; Moretti et al., 2020; Chemello et al., 2021; Koblan et al., 2021).
Tabla 1. Genes con evidencia publicada de eficacia para la edición del genoma en estudios preclínicos con animales de gran tamaño o en ensayos clínicos
| Gen | Enfermedad para la que el gen es una diana terapéutica | Especies | Referencias |
| PCSK9 | Hipercolesterolemia familiar | Monos | Wang et al., 2018; Wang et al., 2021; Rothgangl et al., 2021; Musunuru et al., 2021 |
| ANGPTL3 | Hipercolesterolemia familiar | Monos | No publicado, reportado en conferencias |
| TTR | Amiloidosis transtiretina | Monos, Humanos | Gillmore et al., 2021 |
| DMD | Distrofia muscular de Duchenne | Perros, Cerdos | Amoasii et al., 2018; Moretti et al., 2020 |
| KLKB1 | Angioedema hereditario | Monos | No publicado, reportado en conferencias |
Resulta muy prometedor el ensayo clínico de fase I que ya se está realizando en el que se utiliza la edición del genoma para el tratamiento de la polineuropatía amiloide transtiretina, con el gen TTR (que codifica la transtiretina, una proteína de transporte de tiroxina y retinol) como diana en el hígado (Finn et al., 2018). Los resultados provisionales de la administración a seis pacientes de una dosis única de LNP que contenían el ARNm de Cas9 y un ARN guía sintético que tenía como diana el gen TTR, han mostrado una reducción del 96% de los niveles de transtiretina en sangre, un mes después del tratamiento (Gillmore et al., 2021). A pesar de que todavía se tiene que determinar la cantidad de TTR editado y la durabilidad del efecto terapéutico, y de que este ensayo clínico está enfocado en el tratamiento de la polineuropatía, el alto grado de reducción de transtiretina observado también podría predecir un beneficio para los pacientes con cardiomiopatía causada por la amiloidosis transtiretina.
Es probable que pronto empiecen ensayos clínicos con tratamientos de edición del genoma para otras enfermedades cardiovasculares. Notablemente, casi todo el trabajo en el que se han basado estos ensayos clínicos se ha realizado en la última década. Podemos esperar, sin ninguna duda, que la próxima década vea el mismo progreso extraordinario en el desarrollo de estrategias de edición del genoma, para el beneficio final de los pacientes de todo el mundo.
Conflictos de intereses
El autor es asesor y tiene acciones en las empresas Verve Therapeutics y Variant Bio.