
La leucemia linfocítica crónica (LLC) es una enfermedad extremadamente heterogénea, tanto por sus alteraciones moleculares como por la presentación clínica y la evolución de la enfermedad, que deriva de la expansión clonal de los linfocitos B maduros y cuya acumulación provoca un desequilibrio entre la muerte y la proliferación celular. En los últimos años, el tratamiento de la LLC ha experimentado una revolución con la aparición de los agentes orales dirigidos, que ha ido acompañada de una mejora en la supervivencia y en la calidad de vida de los pacientes. Este cambio de paradigma también afecta al valor de los biomarcadores pronósticos y/o predictivos y a los modelos pronósticos que orientan las opciones terapéuticas y que derivan de la integración de las características clínicas de cada paciente con sus alteraciones genéticas (González-Gascón-y-Marín et al., 2021).
Numerosos estudios multicéntricos han permitido una mejor comprensión de la patogénesis de la LLC al revelar una gran cantidad de alteraciones genéticas, muchas de ellas recurrentes, que afectan a diversas vías y procesos celulares con potencial relevancia clínica (Puente et al., 2015; Landau et al., 2015). Asimismo, estos estudios han permitido perfilar la evolución clonal que subyace a la enfermedad, definida por la adquisición o selección sucesiva de clones tumorales que transforman el diagnóstico de una enfermedad inicialmente indolente y estable hasta estados más agresivos que predisponen a la resistencia al tratamiento y, finalmente, a la transformación a Síndrome de Richter.
El gen más relevante en la LLC es TP53, biomarcador de mal pronóstico, cuya incidencia al diagnóstico supone del 4 al 8%, alcanzando 10 – 12% en el momento de comenzar la primera línea de tratamiento, 40% en las recaídas y 50 – 60% en la evolución a Síndrome de Richter (Bosch & Dalla-Favera., 2019). Las alteraciones en este gen se asocian con una escasa respuesta a la quimioinmunoterapia convencional, una supervivencia global más corta y un menor tiempo de progresión en los pacientes en observación con alto riesgo de transformación (Malcikova et al., 2018; Brown et al., 2018; Malcikova et al., 2014; Gonzalez et al., 2011). TP53 puede estar alterado en la LLC por deleciones, mutaciones o por una combinación de ambas, siendo esto último lo más frecuente. Por ello, las guías internacionales recomiendan evaluar tanto la presencia de deleciones de la región en la que se localiza el gen TP53 por FISH (del(17p13)), como la presencia de variaciones genéticas mediante la secuenciación de la región codificante del gen (Eichhorst et al., 2020; Hallek et al., 2018; Baliakas et al., 2015). En TP53 son igualmente importantes los subclones (variantes con una frecuencia alélica, VAF, < 10%) que subyacen a la enfermedad, presentes en una fracción significativa de pacientes con LLC, y con el mismo impacto pronóstico desfavorable que los defectos clonales del gen. Estos subclones también anticipan el desarrollo de un fenotipo quimioresistente entre los pacientes que requieran tratamiento, y que, según los estudios, conlleva finalmente a la expansión del subclon minoritario hacia un clon refractario predominante (Campo et al., 2018; Rossi et al., 2014).
Las mutaciones de NOTCH1 se observan en el 10% y el 15% de la LLC en el momento del diagnóstico, aumentando su frecuencia aproximadamente un 20% en los pacientes que evolucionan o recaen. Las mutaciones, localizadas principalmente en el exón 34 o en la región 3’ UTR, identifican a un grupo de pacientes con enfermedad de riesgo intermedio y con mayor probabilidad de transformación a Síndrome de Richter. Asimismo, las mutaciones en este gen se asocian con IGHV no mutado, con la trisomía del cromosoma 12 y con niveles bajos de CD20, por lo que los pacientes portadores de mutaciones no parecen beneficiarse de la adición de un anti-CD20 a la quimioterapia (González-Gascón-y-Marín et al., 2021; Moia et al., 2020; Patriarca et al., 2020; Bosch & Dalla-Favera, 2019).
Las mutaciones en SF3B1 se observan en menos de un 10% de las LLC de reciente diagnóstico y aumentan hasta un 20-30% conforme progresa la enfermedad. Generalmente están representadas por cambios de nucleótidos en una región hotspot, siendo la variante K700E la más frecuente (~50%) y permiten identificar a un grupo de pacientes con riesgo intermedio. Estas mutaciones se asocian con IGHV no mutado y tienden a coexistir con la deleción 11q o con mutaciones en el gen ATM (González-Gascón-y-Marín et al., 2021; Moia et al., 2020; Patriarca et al., 2020; Bosch & Dalla-Favera, 2019).
Las deleciones en 11q23 siempre incluye al gen ATM pero además pueden presentarse mutaciones concomitantes en el otro alelo del gen, distribuidas en toda la secuencia codificante, sin hotspots definidos, presentes en aproximadamente un 10 % de los pacientes en el momento del diagnóstico y en alrededor de un 15% durante la progresión de los pacientes que requiere una primera línea de tratamiento. Estas alteraciones identifican a un grupo de pacientes de riesgo intermedio. Asimismo, el gen BIRC3 que se encuentra ubicado cerca del locus de ATM, se observa alterado en el 80% de los pacientes con deleciones en 11q y confiere un pronóstico desfavorable (González-Gascón-y-Marín et al., 2021; Moia et al., 2020; Patriarca et al., 2020; Bosch & Dalla-Favera, 2019).
Las mutaciones de MYD88 ocurren en alrededor de un 3% de la LLC, se asocian a pacientes jóvenes y a las mutaciones de IGHV. Los pacientes con estas mutaciones presentan una supervivencia similar a la población normal (González-Gascón-y-Marín et al., 2021; Moia et al., 2020; Patriarca et al., 2020; Bosch & Dalla-Favera, 2019).
La introducción de fármacos dirigidos que inhiben la señalización de BCR está cambiando la genética de la enfermedad, y ha revelado la adquisición de mutaciones en los genes de la vía BCR, incluyendo mutaciones de BTK dirigidas al codón 481 y que afectan al sitio de unión del fármaco o mutaciones de ganancia de función en PLCG2. Estas dos mutaciones se encuentran en el 85% de las LLC resistentes a este tratamiento y se detectan hasta 15 meses antes de la recaída clínica (González-Gascón-y-Marín et al., 2021; Moia et al., 2020; Patriarca et al., 2020; Bosch & Dalla-Favera, 2019; Hamasy et al., 2017).
Junto a las alteraciones que afectan a procesos celulares como la apoptosis o la respuesta al daño del DNA, existen aberraciones en la vía de señalización BCR que son muy relevantes para estos pacientes. Veinte años después de las primeras publicaciones, existe un consenso de que el estado mutacional de IGHV es una piedra angular importante para una estratificación precisa del riesgo y la toma de decisiones terapéuticas en los pacientes con LLC (Giudice et al., 2019). Las alteraciones presentes en IGHV permiten dividir a los pacientes con LLC en dos grandes grupos según el porcentaje de homología respecto a la línea germinal con un impacto pronóstico independiente de otros factores bien conocidos como son la del17p o la clasificación de Binet (Sutton et al., 2017). Aquellos casos con ≥98% de homología son considerados no mutados (U) y se asocian con una peor evolución clínica, estadios de la enfermedad más avanzados, una morfología celular atípica, reordenamientos de mal pronóstico y resistencia al tratamiento, mientras que los casos con homología <98% considerados mutados (M), presentan un mejor pronóstico. Simultáneamente, se ha observado que una proporción de pacientes con LLC se puede clasificar en 19 subgrupos (subsets) o estereotipos de acuerdo a la similitud de la secuencia CDR3 de las IgH con un perfil de reconocimiento antigénico similar. Las últimas guías internacionales de la European Research Initiative on CLL (ERIC) han reconocido el valor pronóstico de estos subgrupos, principalmente de los subgrupos # 1, # 2 y # 8, asociados a un comportamiento clínico más agresivo, y del subgrupo # 4 que incluye a pacientes con LLC más indolente que rara vez precisan de tratamiento (Stamatopoulus et al., 2017; Baliakas et al., 2015; Malcikova et al., 2014).
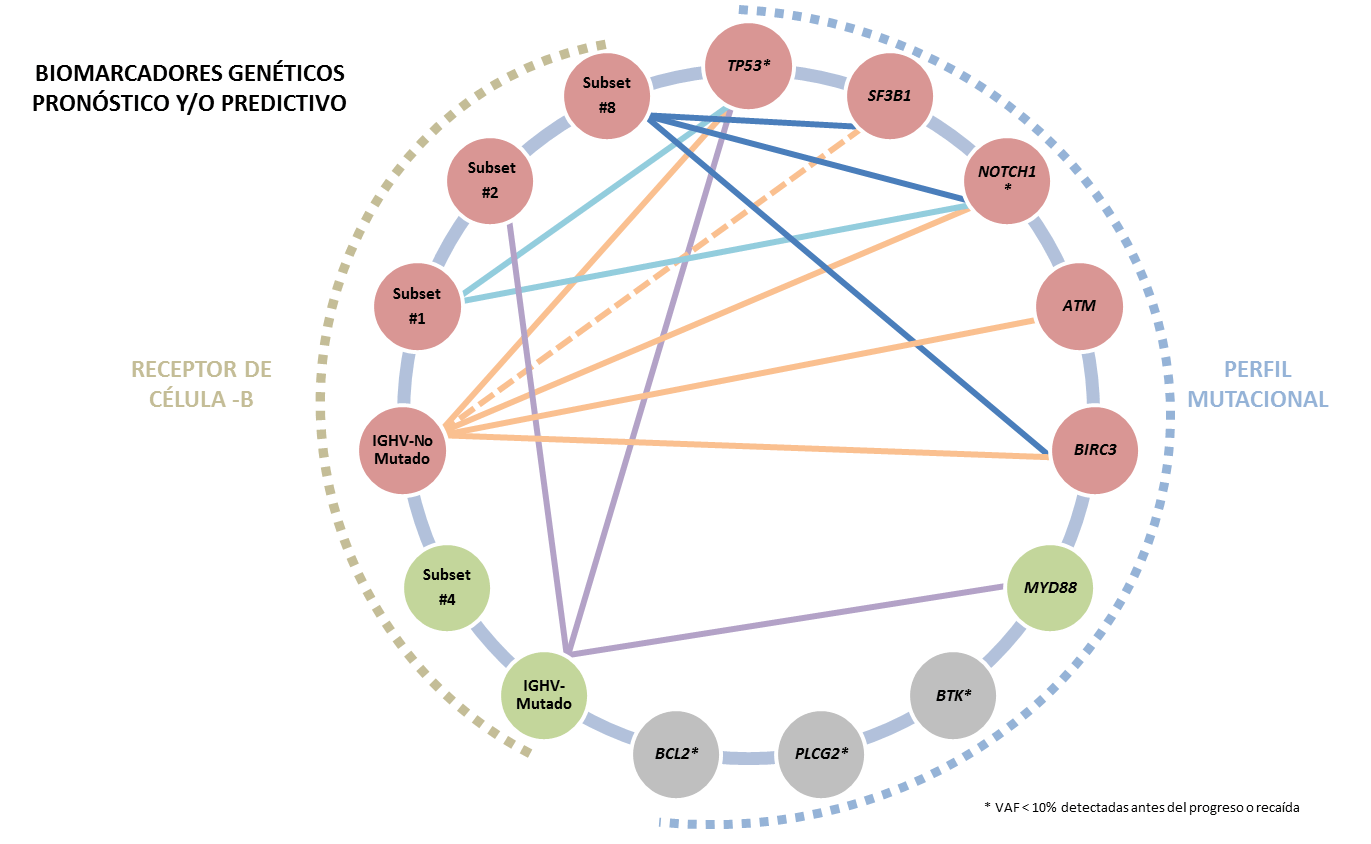
Todas estas alteraciones moleculares se comportan siguiendo un patrón de interacción significativo de incidencia, coocurrencia, y/o exclusividad mutua, permitiendo establecer un algoritmo pronóstico para estos pacientes atendiendo a sus panorama inmunogenético (Figura 1) (Tausch et al., 2020; Giudice et al., 2019; Stamatopoulus et al., 2017; Baliakas et al., 2015). Para muchas de ellas existe un arsenal terapéutico que incluye tanto estrategias de quimioinmunoterapia como fármacos biológicos con eficacias directamente relacionadas con las características moleculares específicas del paciente. Del mismo modo, la enfermedad residual mínima también está emergiendo como un biomarcador relevante con posibles implicaciones clínicas todavía por explorar (Moia et al., 2021; Bosch & Dalla-Favera, 2019).
Sin embargo, pese a todos estos hallazgos y avances, las guías internacionales son muy claras. Para el conjunto internacional, no está indicado actualmente realizar ningún estudio molecular al diagnóstico de una LLC siempre que no requiera tratamiento, aunque para European Society for Medical Oncology (ESMO) el estudio de IGHV estaría justificado para la evaluación de su score pronóstico. Antes del inicio de tratamiento, generalmente administrado al 66% del total de pacientes con LLC, es imprescindible estudiar, al menos, el estado mutacional de TP53 y de IGHV, siendo especialmente relevante documentar el estatus de TP53. Sin embargo, todavía existe controversia en cuanto a la técnica de secuenciación que se debe realizar para el estudio molecular de estos pacientes (Eichhorst et al., 2020; Zalcberg et al., 2020; Hallek et al., 2018; Baliakas et al., 2015).
Las guías internacionales vigentes recomiendan la metodología de secuenciación por Sanger como método estándar para la determinación de las hipermutaciones somáticas del gen IGHV, sin embargo, existe una creciente demanda de cambios en la práctica clínica que conlleve el uso de secuenciación masiva (NGS). Aunque ambos métodos requieren de una gran habilidad y experiencia del usuario con software específico y la interpretación de los datos, con NGS es más sencillo el análisis de las lecturas pese a que la interpretación es mucho más compleja y no está estandarizada. Además la NGS permite la identificación de múltiples reordenamientos productivos de IGHV que no están relacionados clonalmente y que han sido descritos en un cuarto de los pacientes con LLC, manifestándose por tanto la necesidad de redefinir los límites de lo que constituye un clon molecular detectado por NGS (Gupta et al., 2020; Davi et al., 2020; Stamatopoulos et al., 2017). Sin embargo, esta metodología puede llevar a una distorsión de la representación del clon fruto de problemas técnicos como puedan ser, por ejemplo, el sesgo de amplificación y los problemas de cuantificación. Por tanto, queda por determinar si esta variación es un artefacto de la NGS o, de hecho, refleja la verdadera diversidad biológica intraclonal resultante de las hipermutaciones somáticas y cuya importancia clínica todavía requiere de más estudios que impliquen a un mayor número de pacientes (Gupta et al., 2020; Davi et al., 2020; Crombie J et al., 2017).
Actualmente se están realizando numerosos esfuerzos para estandarizar el amplio abanico de herramientas genómicas empleadas en la determinación del perfil mutacional de los pacientes con LLC, que, según las guías, debe centrarse principalmente en TP53. Esta validación está liderada por el ERIC y pretende destacar los parámetros críticos (cobertura, la sensibilidad y reproducibilidad) que los usuarios deben tener en cuenta al introducir NGS en el laboratorio, así como los genes que debe cubrir el análisis (Wiestner, 2021; Sutton et al., 2021). Tras analizar 48 muestras pre-caracterizadas con 3 métodos de secuenciación distintos, en un mismo equipo de secuenciación y con los análisis bioinformáticos centralizados, logran una concordancia del 94% para la detección de variantes con un VAF > 5%, observándose las discrepancias en las variantes subclonales. Por ello, la gran discusión sigue siendo determinar cuál es el límite de VAF en el que se deben de informar las variantes detectadas (Sutton et al., 2021). En este sentido, un estudio multicéntrico llevado a cabo dentro de este mismo grupo cooperativo internacional situaría el dintel en un VAF ≥ 2% (Pavlova et al., 2020), pero continúan siendo necesarios más estudios que nos permitan elucidar los límites que nos permitan trasladar todos estos datos a la práctica clínica diaria del laboratorio de biología molecular.
Con todo esto, dadas las dificultades técnicas, ¿realmente necesitamos conocer las mutaciones de baja frecuencia? En nuestra opinión sí, al menos, para los genes con mayor implicación diagnóstico, pronóstico y/o predictivo, donde las variantes de bajo nivel pueden ser clínicamente relevantes. Esto es cierto no sólo para TP53, cuyos clones sabemos que se expanden en el momento de la recaída, sino también para mutaciones específicas en BTK o PLCG2 y BCL2 que se asocian con la progresión de la enfermedad con inhibidores de BTK o venetoclax, respectivamente, y que pueden detectarse con una baja VAF muchos meses antes de una progresión clínica manifiesta (Tausch et al., 2020). Si bien la presencia de estas mutaciones no justifica cambios inmediatos en el tratamiento, pueden identificar pacientes que podrían beneficiarse de una observación más cercana. Sin embargo, es esencial la estandarización tanto de los aspectos técnicos como de la interpretación para alcanzar una mayor sensibilidad y garantizar una detección consistente y precisa de variantes, principalmente, de baja frecuencia. Asimismo, de momento, es importante considerar la infiltración tumoral de los pacientes con variantes subclonales y realizar estudios evolutivos, aunque su papel continúe siendo controvertido.
Sabemos que las herramientas de NGS para la medicina de precisión en estos pacientes están listas, pero ¿estamos preparados para su traslación? Los datos sobre la información pronóstico y o predictiva de los biomarcadores genéticos para diferentes tipos y líneas de tratamiento son sólidos y deberían ser suficientes. Por ello, la implantación del estudio del perfil molecular completo de los pacientes con LLC por NGS es esencial ya que no sólo proporciona información valiosa para establecer su pronóstico, sino también para establecer marcadores predictivos que guían a los pacientes hacia un amplio abanico de terapias más prometedoras, con distintos mecanismos de acción y en constante crecimiento.


