
INTRODUCCIÓN
Las neoplasias mieloides (NM) son un grupo de enfermedades clonales de la médula ósea que incluyen diversas entidades como la leucemia mieloide aguda (LMA), los síndromes mielodisplásicos (SMD), los síndromes mielodisplásicos/mieloproliferativos (SMD/NMP) y las neoplasias mieloproliferativas (NMP).
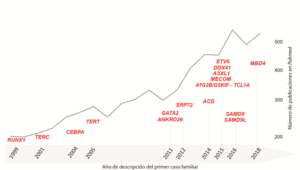
Las NM se presentan principalmente como enfermedades esporádicas en la población de edad avanzada. Sin embargo, recientemente se han publicado casos de agregación familiar con debut de NM en adultos jóvenes que sugieren un componente hereditario. Otro escenario es el hallazgo incidental de variantes con una frecuencia alélica en torno al 50% en genes asociados a predisposición a NM al secuenciar tejido tumoral de pacientes con SMD/LMA (Bannon et al., 2016). El avance en las técnicas de secuenciación de genes ha permitido, en un número cada vez mayor de estos casos, la detección y confirmación de variantes de origen germinal asociadas a predisposición a NM, lo que indica que el número de NM de componente hereditario ha podido estar subestimado hasta ahora (Churpek et al., 2013) (Figura 1).
Estos síndromes hereditarios están adquiriendo tal importancia que la revisión de 2016 de la OMS ha incluido una sección sobre predisposición germinal a síndromes hematológicos (Tabla 1). De hecho, esta última actualización pone énfasis en que el diagnóstico debe incluir el estudio de posibles anormalidades genéticas subyacentes o un síndrome de predisposición mieloide (Arber et al., 2016).
Tabla 1. Clasificación de las NM de predisposición germinal (modificado de Arber et al., 2016).
NM, neoplasias mieloides; mg, mutaciones germinales.
| Clasificación de NM de predisposición germinal | ||
|
NM de predisposición germinal sin alteraciones preexistentes o disfunción de órganos |
NM de predisposición germinal y alteraciones plaquetarios preexistentes |
NM de predisposición germinal y disfunción de otros órganos |
| LMA con mg en CEBPA | NM con mg en RUNX1 | NM con mg en GATA2 |
| NM con mg en DDX41 | NM con mg en ANKRD26 | NM asociadas a síndromes de fallo medular (Anemia de Fanconi, Disqueratosis Congénita; Anemia de Diamond-Blackfan; Síndrome de Shwachman-Diamond y Neutropenia Congénita Severa) |
| NM con mg en ETV6 | NM asociadas a alteraciones de
teloméricas |
|
| LMMJ asociada con neurofibromatosis o Síndrome de Noonan | ||
| NM asociadas a síndrome de Noonan | ||
| NM asociadas a síndrome de Down | ||
Sin embargo, el diagnóstico de NM con predisposición genética es complicado debido a la diversidad de pedigrís específicos de cada familia, la variabilidad en la edad de aparición, la expresión variable y la penetrancia incompleta. Todos estos factores apuntan a que las variantes germinales requieren la acumulación de variantes somáticas adicionales para llegar al debut de la NM. Además, el diagnóstico genético se ve complicado por la existencia de un solapamiento entre variantes patológicas que pueden presentarse tanto de forma esporádica como hereditaria (Furutani et al., 2017).
Los genes implicados en las NM, presentan un perfil clínico, biológico y molecular diferente, que hace imperativo estudiar cada caso de manera individual. En esta revisión recopilamos brevemente las características clínicas y patológicas de las distintas categorías de predisposición germinal a SMD/LMA, centrándonos en los genes descritos más recientemente (Tabla 2), y haciendo alusión a otros genes y síndromes que predisponen al desarrollo de NM.
Tabla 2. Características clínicas y moleculares de los genes asociados a neoplasias mieloides de componente hereditario.
AA, anemia aplásica; AD, autosómica dominante; CNV, copy number variant: cambio en el número de copias; DLBCL, linfoma difuso de células B grandes; frameshift, con cambio de marco de lectura; LMMJ, leucemia mielomonocítica juvenil; LLC, leucemia linfática crónica; LH, linfoma Hodgkin; missense, con cambio de sentido; nonsense, con pérdida de sentido; PV, policitemia vera; sFMO, síndrome de fallo medular; SMP, síndrome mieloproliferativo; TE, trombocitopenia esencial.
|
Gen |
Posición cromosómica |
Efecto en la proteína |
Transcrito |
Variantes más recurrentes |
Herencia |
Penetrancia |
Desórdenes hematológicos |
Otras Complicaciones |
Nº de familias descritas |
Edad |
Referencias |
| ACD | 16q22.1 | Deleciones, missense | NM_001082486 | c.508_510delAAG0;p.K170del
c.1471C>A;p.P491T |
AD | Incompleta | sFMO, DC, AA | Síndrome Hoyeraal-Hreidarsson | 2 | Muy variable (anticipación) | Guo et al., 2016 |
| ANKRD26 | 10p12.1 | Deleciones, missense | NM_014915 | Región 5′ – UTR,
Exones 1 y 2 |
AD | ≈100% | SMD, LMA, LLC, LMC, LMMC | No descritas | 45 | 1-84 | Botero et al., 2018 |
| ASXL1 | 20q11.21 | Missense | NM_015338 | c.2957A > G;p.N986S c.1205 G>A;p.R402E |
No establecida | Incompleta | SMD, LMA, LLA, LNH | Síndrome Bohring Opitz | 2 | Muy variable (anticipación) | Hamadou et al., 2016; Seiter et al., 2018 |
| ATG2B/GSKIP – TCL1A | 14q32.2 | Duplicación | – | Duplicación | AD | Incompleta | SMP, TE, mielofibrosis, LMA, LMMC | No descritas | 5 | 22-76 | Saliba et al., 2015; Babushok et al., 2018 |
| CEBPA | 19q13.11 | Frameshift, nonsense | Región N-terminal | AD | ≈100% | LMA | No descritas | 20 | 2-46 | Tawana et al., 2017 | |
| DDX41 | 5q35.3 | Frameshift, missense, splicing | NM_016222.2 | c.415_418dupGATG; p.D140Gfs*2 | AD | Incompleta | SMD, LMA, LMMC, LNH, LH, MM | No descritas | 20 | 40-85 | Cheah et al.,2017; Maciejewski et al., 2017 |
| ETV6 | 12p13.2 | Frameshift, missense, nonsense, splicing, CNV | NM_001987 | c.1106G>A;p.A369Q
c.641C>T;p.A399C c.641C>T;p.P214L |
AD | Incompleta | SMD,LMA, MM, PV, LMMC,SMP | Riesgo de tumores sólidos, desordenes autoinmunes | 7 | 3-81 | Zhang et al., 2015;
Hock et al., 2017 |
| GATA2 | 3q21.3 | Deleciones, frameshift, missense, nonsense, splicing | NM_032638 | c.1061C>T;p.T354M | AD/de novo | ≈100% | sFMO, SMD, LMA | Linfedemas, inmunodeficiencia, pérdida de audición, tumores solidos | No establecido | <1-78 | Gao et al., 2014;
Wlodarski et al., 2017 |
| MBD4 | 3q21.3 | Deleciones, frameshift | NM_003925 | c.1699_1701delCAT;p.H567del c.939_940insA; p.V314RfsTer13 |
No establecida | No establecida | LMA | Tumores sólidos | 2 | 30-34 | Sanders et al., 2018 |
| MECOM/ MDS1-EVI1 | 3q26.2 | Deleciones, missense | NM_001105078 | c.2296T>G;p.C766G | AD/de novo | Incompleta | sFMO, LMA | RUSAT2, malformación de órganos, tumores sólidos, pérdida de audición | No establecido | 0-73 | Bluteau et al., 2018; Ripperger et al., 2018 |
| RUNX1 | 21q22.12 | Deleciones, duplicaciones, inserciones, frameshift, missense, nonsense, splicing | NM_001754 | Domino RUNT | AD | Incompleta | FPD, SMD, LMA,LLA,LNH, LLC, DLBCL, mielofibrosis, linfosarcoma | Eczema | 70 | 6-75 | Hayashi et al., 2017 |
| SAMD9 | 7q21.2 | Missense | NM_017654 | c.3406G>C;p.E1136Q
c.2026A>G;p.K676E |
AD/de novo | Incompleta | sFMO, SMD, LMA | MIRAGE, atrofia de órganos | 19 | Infancia | Davidsson et al., 2018; Wong et al., 2018 |
| SAMD9L | 7q21.2 | Missense | NM_152703 | c.2640C>A;p.H880Q c.2956>T;p.R986C c.3587G>C;p.C1196S c.2672T>C;p.I891T |
AD | Incompleta | sFMO, SMD, LMA, LMMJ | ATXPC, atrofia de órganos | 8 | 1-56 | Davidsson et al., 2018; Wong et al., 2018 |
| SRP72 | 4q12 | Deleciones, missense | NM_006947 | c.1064_1065del;p.T355Kfs*19 c.620G>A;p.R207H |
AD | No establecida | sFMO, AA, SMD | Pérdida de audición | 2 | 11-76 | Kirwan et al., 2012 |
| TERC | 3q26.2 | Deleciones, duplicaciones, framesehift, missense, nonsense | NR_001566 | Desconocida | AD | Incompleta | sFMO, AA, SMD, LMA | Fibrosis pulmonar y hepática, tumores sólidos | No establecido | Muy variable (anticipación) | Kirwan et al., 2009 |
| TERT | 5p15.33 | Deleciones, duplicaciones, framesehift, missense, nonsense | NM_198253.2 | Desconocida | AD/AR | Incompleta | sFMO, AA, SMD, LMA | Fibrosis pulmonar y hepática, tumores sólidos | No establecido | Muy variable (anticipación) | Calado et al., 2009; Kirwan et al., 2009; Savage et al., 2010 |
NEOPLASIAS MIELOIDES DE PREDISPOSICIÓN GERMINAL SIN ALTERACIONES PREEXISTENTES NI DISFUNCIÓN DE OTROS ÓRGANOS
LMA con mutaciones germinales en CEBPA
CEBPA (CCAAT enhancer binding protein alpha) es un gen con un único exón que codifica para un factor de transcripción clave en la diferenciación de linaje mieloide, perteneciente a la familia bZIP. Su expresión es característica en células mieloides tempranas e interviene en la diferenciación granulocítica y monocítica (Radomska et al., 1998). Las mutaciones en CEBPA se han descrito entorno al 10% de los pacientes con LMA. (Fasan et al., 2014)
La LMA familiar con alteraciones en CEBPA es de tipo autosómica dominante con penetrancia cercana al 100%. Habitualmente los pacientes heredan una copia mutada del gen en el extremo N-terminal. El desarrollo de leucemia se asocia a la adquisición de una alteración bialélica en el extremo C-terminal (Taskesen et al., 2011).
Las formas esporádica y hereditaria de la enfermedad comparten características incluyendo una analítica sanguínea normal, cariotipo normal, presencia de bastones de Auer y expresión aberrante de CD7 en blastos (Godley, 2014).
El pronóstico general es favorable en pacientes con una variante bialélica, con tasas de supervivencia a 5 años cercanas al 70%, por lo que el trasplante alogénico no es mandatorio. Sin embargo, a pesar de que los pacientes con LMA con alteraciones en CEBPA en la línea germinal responden bien a la quimioterapia, son propensos al desarrollo de clones malignos diferentes a los del diagnóstico (Pabst et al., 2008) .
NM con mutaciones germinales en DDX41
DDX41 (DEAD-box helicase 41) codifica para una helicasa de ARN con funciones en el splicing del ARN mensajero, la inmunidad innata y la biogénesis ribosómica. Su papel en la hematopoyesis y la leucemogénesis no está bien definido (Polprasert et al., 2015; Cheah et al., 2017).
Las NM con alteraciones en DDX41 presentan herencia de tipo autosómica dominante. Fueron descritas por primera vez en 2015 (Babushok et al., 2017). Las alteraciones en DDX41 se encuentran de forma esporádica en aproximadamente el 1,5% de los pacientes con neoplasias mieloides y cerca del 50% de estos, tienen variantes patogénicas en la línea germinal. Es frecuente la detección alteraciones somáticas adicionales de DDX41 en el alelo no mutado en línea germinal (Babushok et al., 2017). Además de mutaciones en DDX41, se dan casos en los que hay una deleción de la región 5q35 que conlleva una expresión haploinsuficiente de este gen (Polprasert et al., 2015).
A diferencia de otros síndromes de predisposición, el desarrollo de leucemia hereditaria asociada a DDX41 debuta a finales de la edad adulta, lo que dificulta su distinción de los casos originados por variantes somáticas (Lewinsohn et al., 2016).
La mayoría de los pacientes presentan hemogramas y cariotipos normales previos al diagnóstico de la neoplasia. Las entidades típicamente asociadas a mutaciones germinales en DDX41 son SMD, LMA y Leucemia Mielomonocítica Crónica (LMMC), aunque recientemente se ha descrito asociada a otras patologías hematológicas como Leucemia Mieloide Crónica (LMC), linfomas y mieloma múltiple (MM). Una vez se ha desarrollado la neoplasia, es muy característica la aparición de macrocitosis, médula ósea hipocelular y diseritropoyesis (Lewinsohn et al., 2016). En el caso de trasplante, el estudio genético de los posibles donantes es mandatorio ya que puede darse la reaparición de LMA en las células del donante portador (Berger et al., 2017).
NM con duplicaciones en 14q32
Esta citobanda incluye tres genes relacionados con anomalías germinales asociadas a NM: ATG2B (autophagy related 2B), GSKIP (Glycogen synthase kinase-3 beta interacting protein) y TCL1A (T cell leukemia/lymphoma 1A).
En 2015, se publicó un estudio que describía una duplicación de 700 kb en el cromosoma 14 como un factor de predisposición al desarrollo de desórdenes mieloproliferativos con transformación a mielofibrosis o LMA de tipo hereditaria autosómica dominante con penetrancia incompleta. La región duplicada abarca los genes TCL1A, BDKRB1, BDKRB2, ATG2B, GSKIP y AK7. Los autores de ese trabajo llegaron a la conclusión que ATG2B y GSKIP estaban sobreexpresados en las células hematopoyéticas de los pacientes, y por tanto debían ser los responsables del desarrollo de la LMA (Saliba et al., 2015). Sin embargo, dos publicaciones más recientes indican que GSKIP y ATG2B no están implicados en el desarrollo de NM, y apuntan a la duplicación de TCL1A como el potencial impulsor del proceso leucémico (Babushok et al., 2018; Hahn et al., 2017).
Estos trabajos contradictorios hacen imperativo realizar estudios para determinar con exactitud el mecanismo por el cual las duplicaciones de 14q32 confieren predisposición a NM. Hasta entonces, es aconsejable que los test diagnósticos cubran la región 14q32 completa, sin limitarse únicamente a GSKIP y ATG2B.
Los pacientes con LMA asociada a duplicación en 14q32 debutan con cariotipo complejo y alteraciones moleculares adicionales. El pronóstico es desconocido (Saliba et al., 2015).
NEOPLASIAS MIELOIDES CON PREDISPOSICIÓN GERMINAL Y TRASTORNOS PLAQUETARIOS PREEXISTENTES
NM con mutaciones germinales en RUNX1
RUNX1 (runt related transcription factor 1) codifica una subunidad de un factor de transcripción heterodimérico que controla la expresión de genes esenciales para la hematopoyesis. Las alteraciones germinales se asocian a desórdenes plaquetarios familiares con predisposición al desarrollo de NM, con herencia autosómica dominante y penetrancia variable (Song et al., 1999). Se han identificado hasta 13 pedigríes diferentes, incluyendo alteraciones de tipo missense, nonsense, frameshift, de splicing, deleciones, duplicaciones, inserciones y una gran eliminación intragénica (Béri-Dexheimer et al., 2008).
Los pacientes con alteraciones en RUNX1 presentan una diferenciación hematopoyética deficiente, un descenso en el número de progenitores hematopoyéticos y alteraciones en la diferenciación de los megacariocitos (Sakurai et al., 2014). Es característica una historia de trombocitopenia leve a moderada y agregación plaquetaria defectuosa, aunque también pueden no manifestar signos clínicos. El desarrollo neoplásico en individuos portadores de una alteración germinal en RUNX1 se asocia a la adquisición de una segunda mutación en el alelo no mutado en línea germinal, o bien a mutaciones somáticas en otros genes, como CDC25C, CBL, FLT3, KRAS, TP53, SRSF2, SF3B1, TET2, y DNMT3A (Bellissimo et al., 2017)
Análisis citogenéticos han descrito casos de trisomía 21, monosomía 5, deleción en 5q, deleción en 7q en neoplasias con mutaciones germinales en RUNX1 (Preudhomme et al., 2009). Además, se han publicado casos asociados a neoplasias linfoides en el contexto de un desorden plaquetario familiar (Kanagal-Shamanna et al., 2017).
Debido a la heterogeneidad genética y la variabilidad en las manifestaciones clínicas, los datos relacionados con el manejo y el pronóstico de la enfermedad son limitados.
Trombocitopenia asociada a ANKRD26
ANKRD26 (ankyrin repeat domain 26) se asocia a trombocitopenia tipo 2, una forma rara de trombocitopenia hereditaria de tipo autosómica dominante con penetrancia alta (Noris et al., 2015).
La mayoría de las alteraciones germinales descritas se localizan en el extremo 5’ del gen (UTR, y exones 1 y 2) y consisten en sustituciones de un solo nucleótido. Se cree que estas llevan al aumento de los niveles de expresión de ANKRD26, lo que potenciaría la señalización de la vía MAPK/ERK, dando lugar a una génesis pro-plaquetaria deficiente por parte de los megacariocitos. Las mutaciones en el resto de la región codificante del gen son menos frecuentes (Bluteau et al., 2014).
Los pacientes con mutaciones en ANKRD26 se caracterizan por presentar trombocitopenia moderada con plaquetas de tamaño normal, sangrados espontáneos leves; un 10% de estos pacientes desarrollan una NM (30 veces más predisposición que la población sana); de forma menos frecuente, se han descrito casos de LMC, LMMC y Leucemia Linfática Crónica (LLC) asociados a las mutaciones en este gen. El pronóstico es incierto (Brown et al., 2017).
Trombocitopenia asociada a ETV6
ETV6 (ETS variant 6) es un factor de transcripción que participa en la regulación de la hematopoyesis, en el mantenimiento de la red vascular, y presenta función de supresor tumoral (Feurstein et al., 2017). La trombocitopenia familiar tipo 5 asociada a ETV6 es un síndrome autosómico dominante con penetrancia variable según el tipo de mutación en la línea germinal (Hock et al., 2017) .
La mayoría de las alteraciones germinales descritas en este gen son de tipo missense. Estas tienen un efecto dominante negativo, que provoca un defecto en su localización nuclear. Como consecuencia se reduce la expresión de genes asociados a plaquetas (Geyer, 2018).
Los pacientes afectados presentan trombocitopenia variable, con plaquetas de tamaño normal, tendencia leve a moderada de sangrados, y macrocitosis eritroidea. Las biopsias de médula ósea revelan megacariocitos pequeños hipolobulados, hipogranulación en las células mieloides y diseritropoyesis leve (Hock et al., 2017). Además, las mutaciones en línea germinal de ETV6 confieren una predisposición a desarrollar diferentes neoplasias asociadas a un fallo de médula ósea: SMD, LMA, LMMC, MM y leucemia linfoblástica aguda (LLA), además de un riesgo aumentado a desarrollar tumores sólidos. El pronóstico es incierto (Geyer, 2018).
NEOPLASIAS MIELOIDES CON PREDISPOSICIÓN GERMINAL ASOCIADAS A OTROS SÍNDROMES Y A ALTERACIONES EN OTROS ÓRGANOS
NM con mutaciones germinales en GATA2
GATA2 (GATA binding protein 2) es un factor de transcripción de la familia de dedos de zinc que participa en la regulación de la hematopoyesis, la autoinmunidad y la inflamación. Además, interviene en el desarrollo linfático vascular (Spinner et al., 2018).
Las alteraciones germinales en GATA2 se identificaron originalmente en 2010 en 4 síndromes aislados: el síndrome de deficiencia de GATA2, el síndrome monoMAC (OMIM 137295), NM familiares, el síndrome de Emberger (OMIM 614038). Debido al solapamiento de las características en estos desórdenes, se decidió reconocer las alteraciones germinales de este gen como una sola entidad con manifestaciones pleiotrópicas y un riesgo elevado de desarrollar NM (75%) (Dickinson et al., 2018). Las mutaciones en la línea germinal de GATA2 provocan la pérdida de función del alelo mutado y, por tanto haploinsuficiencia, lo que lleva a la pérdida de células madre hematopoyéticas y a un desarrollo linfático defectuoso (Ganapathi et al., 2018).
Los individuos afectados presentan heterogeneidad fenotípica que comprende linfedemas, inmunodeficiencia, y predisposición a desarrollar NM de tipo hereditario. La progresión a mielodisplasia habi-tualmente se asocia a la adquisición de anomalías citogenéticas, tales como monosomía del cromosoma 7, der(1;7), o trisomía de los cromosomas 8 y 1, y a mutaciones somáticas en ASXL1. Por lo general, la médula ósea muestra hipocelularidad y displasia multilínea. La dismegacariopoyesis es la característica más prominente, observada en el 82% de los casos estudiados (West et al., 2013; Spinner et al., 2018). El pronóstico es pobre, por lo que se recomienda la realización de un trasplante alogénico con la precaución de realizar el estudio genético de los donantes, ya que al igual que en el caso de DDX41, puede darse la reaparición de SMD/LMA en las células del donante portador (Galera et al., 2018).
TELOMEROPATÍAS
Aplasia medular asociada a telomeropatías: TERT y TERC
En el contexto de los síndromes hereditarios de aplasia medular, se han identificado genes causantes de disqueratosis congénita (DC, OMIM 305000) asociados a telomeropatías.
Los telómeros son secuencias repetitivas presentes en ambos extremos de los cromosomas eucariotas, que juegan un papel fundamental en el mantenimiento de la integridad genómica. Los telómeros se acortan con cada división celular debido a la replicación incompleta de los extremos 3′ del ADN y, por lo tanto, son marcadores de envejecimiento celular. El acortamiento de los telómeros es contrarrestado por la acción de la telomerasa. Esta es una enzima compuesta por una transcriptasa inversa codificada por TERT, que utiliza la molécula de ARN específica TERC como molde para alargar el extremo 3′ de la hebra principal añadiendo repeticiones TTAGGG. Los mecanismos de protección de los extremos de los telómeros son necesarios para el mantenimiento del sistema hematopoyético (Armanios, 2009).
Las alteraciones en TERC y TERT (telomerase reverse transcriptase & telomerase RNA component) son causantes del 10% de casos de DC, aproximadamente. Las mutaciones germinales en heterocigosis en estos genes pueden dar lugar a NM de tipo familiar. Cabe destacar que las mutaciones bialélicas en TERT que producen DC autosómica recesiva son generalmente más severas que las monoalélicas autosómicas dominantes (Kirwan et al., 2009).
Los pacientes con alteraciones en TERC o TERT manifiestan los primeros signos de la enfermedad a una edad muy variable, con penetrancia incompleta. Además, se da el fenómeno de anticipación genética, los individuos de generaciones sucesivas dentro de una familia portadora presentan telómeros progresivamente más cortos y una mayor predisposición a desarrollar neoplasias hematológicas a una edad cada vez más temprana (Young, 2012).
Los portadores de mutaciones germinales en TERT o TERC habitualmente presentan un recuento sanguíneo normal con pequeñas variaciones, como volumen corpuscular medio elevado o trombocitopenia, antes de desarrollar la aplasia medular. Habitualmente tienen cariotipo alterado. Algunos pacientes desarrollan fibrosis pulmonar idiopática o cirrosis hepática. La co-ocurrencia de aplasia medular y fibrosis pulmonar se considera predictiva de telomeropatías (Young, 2012).
Aplasia medular asociada a alteraciones germinales de ACD
ACD (shelterin complex subunit and telomerase recruitment factor), codifica para la proteína TPP1, la cual participa en el mantenimiento y en la estabilidad de los telómeros. TPP1 presenta una región en su superficie conocida como TEL patch, que media las interacciones con la telomerasa y es imprescindible para su reclutamiento a los telómeros (Nandakumar et al., 2012).
En el año 2014, se identificaron dos familias con mutaciones germinales en ACD asociadas a alteraciones hematológicas. El tipo de transmisión es autosómica dominante con penetrancia incompleta. En las familias con alteraciones en ACD se da el fenómeno de anticipación genética, relacionado con un aumento en la severidad y el desarrollo temprano de síntomas en generaciones sucesivas. Los pacientes se caracterizan por tener telómeros cortos y aplasia medular. Las alteraciones en ACD también se asocian al síndrome de Hoyeraal-Hreidarsson y al desarrollo de leucemia y tumores sólidos (Kocak et al., 2014).
OTRAS SITUACIONES CON POSIBLE RIESGO DE DESARROLLAR NM
Además, como detalla la revisión de 2016 sobre la clasificación de NM y LMA de la OMS hay otros síndromes asociados a predisposición a NM: NM asociadas a síndromes de fallo medular (Anemia de Fanconi, Anemia de Diamond-Blackfan, Síndrome de Shwachman-Diamond y Neutropenia Congénita Severa), Leucemia mielomonocítica juvenil (LMMJ) asociada con neurofibromatosis o Síndrome de Noonan, NM asociadas a síndromes de Noonan, y NM asociada a síndrome de Down (Tabla 2) (Arber et al., 2016).
Adicionalmente a los genes contemplados en la clasificación de la OMS, existen evidencias bibliográficas de otros genes con variantes en línea germinal en familias en las que dos o más miembros desarrollan NM (ASXL1, SRP72, SAMD9, MBD4, y MECOM/MDS1-EVI1). Todos estos genes presentan aberraciones somáticas causales de NM, y por eso los recogemos en esta revisión, aunque aún es necesario el estudio de más casos para poder establecer una relación robusta entre la presencia de las variantes en línea germinal con el desarrollo de NM.
NM asociadas a mutaciones germinales en ASXL1
ASXL1 (Additional sex comb like 1) se trata de un gen perteneciente al grupo de proteínas Polycomb. La proteína codificada por ASXL1 funciona como un co-activador dependiente de ligando para el receptor del ácido retinoico. Se expresa en la mayoría de células hematopoyéticas y está involucrado tanto en procesos de desarrollo como en contextos de enfermedad, incluyendo la transformación de células normales a tumorales, así como defectos estructurales y enfermedad mental (Gelsi-Boyer et al., 2012).
Las mutaciones germinales en ASXL1 se describieron inicialmente en el Síndrome de Bohring Opitz (OMIM 605039), una enfermedad rara caracterizada por malformaciones múltiples, discapacidad intelectual severa, y corta esperanza de vida. Por otra parte, existen diferentes alteraciones somáticas que involucran a ASXL1 en NM de mal pronóstico, incluyendo SMD, LMA, NMP y leucemia de tipo linfoide (Gelsi-Boyer et al., 2012; Carlston et al., 2017).
Se cree que las mutaciones germinales en ASXL1 también podrían contribuir al desarrollo NM de componente hereditario. En 2015 se identificó una familia en la cual cuatro individuos portaban una mutación missense en línea germinal en ASXL1; dos de ellos desarrollaron un linfoma no Hodgkin (LNH) (Hamadou et al., 2016). En 2018, se publicó un estudio en el que un padre y un hijo portaban también una mutación missense germinal en ASXL1. En esta familia, ambos individuos presentaban SMD con progresión a LMA con cariotipo complejo (Seiter et al., 2018). Se necesitan más investigaciones sobre las alteraciones germinales de ASXL1 para poder establecer la contribución de las mismas al desarrollo de alteraciones hematológicas constitucionales .
Aplasia medular familiar y SMD asociados a mutaciones germinales en SRP72
SRP72 (signal recognition particle 72) codifica la subunidad de 72 kDa de la partícula de reconocimiento de señal (SRP), un complejo ribonucleoproteínico responsable de detener la traducción de proteínas secretoras o extracelulares y dirigirlas al retículo endoplásmico (Kirwan et al., 2012).
Las mutaciones germinales en SRP72 se asocian a aplasia y predisposición al desarrollo de formas hereditarias de SMD de herencia dominante. Los primeros casos descritos fueron en dos familias no relacionadas en el año 2011; individuos de ambas familias presentaban desórdenes hematológicos además de pérdidas de audición o anomalías audiovestibulares (Godley, 2014; Babushok et al., 2017).
Debido a la escasez de casos descritos, se desconoce la incidencia, el riesgo de desarrollar una neoplasia o qué indicaciones clínicas seguir en estas familias.
Monosomía 7 y SMD asociados a mutaciones germinales en SAMD9 y SAMD9L
SAMD9 y SAMD9L (sterile alpha motif domain containing 9 y sterile alpha motif domain containing 9 like) comparten alrededor del 60% de su secuencia. Ambos genes participan en la regulación de la proliferación celular y la apoptosis. Además, SAMD9L interactúa en la respuesta inmunitaria innata frente a infecciones víricas (Davidsson et al., 2018).
Las mutaciones germinales descritas en estos genes son de tipo de ganancia de función, y provocan un aumento del efecto antiproliferativo de los mismos. Es habitual la pérdida total o parcial del cromosoma 7 (que incluye la pérdida del alelo mutado) y la adquisición de mutaciones somáticas aberrantes que revierten el efecto antiproliferativo propiciando la expansión clonal y el desarrollo de NM (Davidsson et al., 2018).
Las alteraciones en SAMD9 parecen asociarse a un fenotipo más grave, que incluye el síndrome MIRAGE (OMIM 617053) caracterizado por mielodisplasia, infecciones, restricción del crecimiento intrauterino, retraso del desarrollo e hipoplasia de órganos no hematopoyéticos. La mayoría de pacientes descritos fallecen en la infancia como consecuencia de hemorragias (Davidsson et al., 2018). Las mutaciones germinales en SAMD9L se relacionan con el síndrome de ataxia-pancitopenia (ATXPC, OMIM 159550), además de con neoplasias hematológicas, tanto en niños como en adultos (Wong et al., 2018).
Neoplasias mieloides con deficiencia en MBD4
MBD4 (methyl-CpG binding domain 4, DNA glycosylase) participa en la estabilidad genómica mediante la prevención de la acumulación de mutaciones en sitios CpG. Interviene en la apoptosis en respuesta al daño en el ADN, represión transcripcional, estabilidad cromosómica, y en el cambio de isotipo de las inmunoglobulinas (Tricarico et al., 2015).
En 2018 se publicó un artículo que describe tres pacientes, (dos de ellos emparentados) con alteraciones en línea germinal en este gen y anomalías citogenéticas, que desarrollaron LMA a edad temprana (West et al., 2014).
Se cree que la deficiencia de MBD4 en la línea germinal promueve la hematopoyesis clonal y el desarrollo de LMA mediante mutaciones adicionales recurrentes en los genes DNMT3A, TP53, ASXL1, IDH1, IDH2 y TET2. Las mutaciones en MBD4 se asocian también a tumores sólidos (Sanders et al., 2018).
RUSAT2 y NM asociadas a mutaciones germinales en MECOM/ MDS1-EVI1
MECOM (MDS1 and EVI1 complex locus) codifica para un factor de transcripción de la familia de dedos de zinc con funciones importantes en el desarrollo y la oncogénesis; además participa en la hematopoyesis y en la renovación de células madre. MECOM codifica diferentes transcritos que producen las isoformas: MDS1, MDS1-EVI1 y EVI1 (Niihori et al., 2015; Germeshausen et al., 2018).
En el año 2015, se describió una familia con mutaciones germinales en MECOM de tipo missense, en el contexto de una sinóstosis radiocubital con trombocitopenia amegacariocítica (RUSAT2, OMIM 616738), un síndrome de fallo medular hereditario caracterizado por trombocitopenia y fusión congénita del radio y cúbito (Niihori et al., 2015).
En el año 2018, se describió otra familia con cuatro individuos portadores de una mutación germinal de tipo missense en este gen. Todos los individuos afectados presentaban manifestaciones clínicas de gravedad variable (RUSAT2, problemas auditivos y alteraciones hematológicas) y la mitad de los pacientes desarrollaron NM (Ripperger et al., 2018).
Hasta el momento, los casos publicados son escasos, por lo que se requieren más estudios para confirmar si MECOM pertenece al grupo de genes cuyas alteraciones germinales contribuyen al desarrollo de NM, investigar la variabilidad clínica y correlaciones genotipo-fenotipo, y establecer el riesgo de desarrollar neoplasias hematológicas (Ripperger et al., 2018).
Por otra parte, recientemente se han identificado variantes patogénicas con predisposición germinal en NMP; los genes afectados incluyen TERT, SH2B3, TET2, ATM, CHEK2, PINT, FG11B, MECOM, TERT, JAK2 y HBSL1-MYB (Bacher et al., 2018). En base a estos datos, parece más que probable que la próxima revisión de la OMS amplíe la lista de NM con predisposición germinal.
CONCLUSIONES
La detección de variantes germinales patogénicas en un paciente diagnosticado de NM, junto al conocimiento detallado de su historia clínica y familiar, puede tener implicaciones en cuanto al cuidado personalizado del individuo, por lo que los datos genéticos y el estudio de su relación con las NM son de gran utilidad clínica. Estos estudios se ven dificultados por el tamaño reducido de las familias en la sociedad actual, la penetrancia incompleta de algunas variantes, la edad tardía de aparición de la enfermedad, y los variados fenotipos asociados a cada gen, por lo que es esencial la colaboración estrecha entre genetistas, investigadores y clínicos, para compartir datos y llegar a conclusiones robustas (Godley, 2014).
Las alteraciones en la línea germinal asociadas a cáncer hereditario constituyen un área de investigación en auge en el campo de las neoplasias hematológicas debido a la mayor accesibilidad a las técnicas de secuenciación masiva y a la mejora de las mismas. Por tanto, es probable que en un futuro cercano se incremente la identificación de genes y variantes involucradas en el desarrollo de neoplasias mieloides hereditarias.
AGRADECIMIENTOS
Investigación financiada por el Gobierno de Navarra, Departamento de Industria, Energía e Innovación (Proyecto DIANA, 0011-1411-2017-000028 y 000030). Los autores quieren expresar su agradecimiento a todo el equipo de CIMA LAB Diagnostics (https://www.unav.edu/web/cimalab/cima-lab-diagnostics/equipo/genetica-de-enfermedades-hematologicas). MFM agradece también financiación de la Asociación Española Contra el Cáncer (AECC, AIO14), y del Instituto de Salud Carlos III (PI16/00159).


