INTRODUCCIÓN
La neurofibromatosis tipo 1 (OMIM#162200), anteriormente conocida como enfermedad de von Recklinghausen, es una enfermedad genética autosómica dominante con una incidencia aproximada de 1 en cada 3.000 nacidos vivos (Giugliano et al., 2019). Se caracteriza por un inicio clínico temprano y manifestaciones predominantemente neurocutáneas, como múltiples manchas café con leche (mayores de 5 mm en prepúberes y de 15 mm en púberes), efélides axilares o inguinales, neurofibromas cutáneos o plexiformes, nódulos de Lisch, glioma de la vía óptica, así como alteraciones esqueléticas, entre ellas displasia del esfenoide o adelgazamiento cortical de huesos largos, con o sin pseudoartrosis (Castle et al., 2023).
Esta condición es causada por variantes patogénicas en el gen supresor tumoral NF1 (OMIM*613113; 17q11.2), que codifica la neurofibromina 1, una proteína activadora de la GTPasa, involucrada como regulador negativo de la vía de señalización RAS/MAPK (Giugliano et al., 2019). Dada su función, los individuos con alteraciones en el gen NF1 presentan mayor susceptibilidad al desarrollo de tumores, predominantemente benignos, aunque también son propensos a neoplasias malignas. Dentro de estas últimas, los sarcomas de tejido blando representan tumores malignos de alto grado (Margo, Johnson y Mancera, 2023), de patogenia no esclarecida que se ha relacionado con factores ambientales como la exposición a radiación ionizante, carcinógenos químicos y virus oncogénicos (Ferrari et al., 2007). Sin embargo, un pequeño porcentaje de casos (<5%) se atribuye a síndromes de predisposición genética, como las RASopatías, que incluyen la neurofibromatosis tipo 1, el Síndrome de Costello (OMIM#218040) y el Síndrome de Noonan (OMIM#163950) (Ji et al., 2019).
Los sarcomas de tejido blando son las neoplasias malignas más frecuentes en edad pediátrica, representando entre el 7% y el 8% de los casos (Margo, Johnson y Mancera, 2023). El rabdomiosarcoma constituye entre el 40% y el 50% de estos sarcomas, distinguiéndose principalmente en dos subtipos: alveolar y embrionario, siendo este último el más frecuente, con aproximadamente el 60% de los casos en la población pediátrica (Ferrari et al., 2007).
La asociación entre neurofibromatosis tipo 1 y rabdomiosarcoma embrionario es infrecuente, con una prevalencia estimada inferior al 1%. En este contexto, el presente manuscrito describe el abordaje clínico, diagnóstico y hallazgos moleculares en una paciente pediátrica con rabdomiosarcoma embrionario asociado a neurofibromatosis tipo 1, representando el primer caso documentado en la literatura médica colombiana.
DESCRIPCIÓN DEL CASO
Paciente femenina de 14 meses de edad, hija de padres no consanguíneos, producto del tercer embarazo a término, nacida a las 40 semanas de gestación con peso de 3.650 gramos y talla de 45 centímetros. El embarazo transcurrió sin complicaciones, sin exposición a teratógenos, infecciones maternas ni uso de técnicas de reproducción asistida.
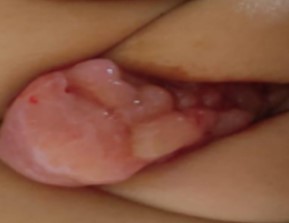
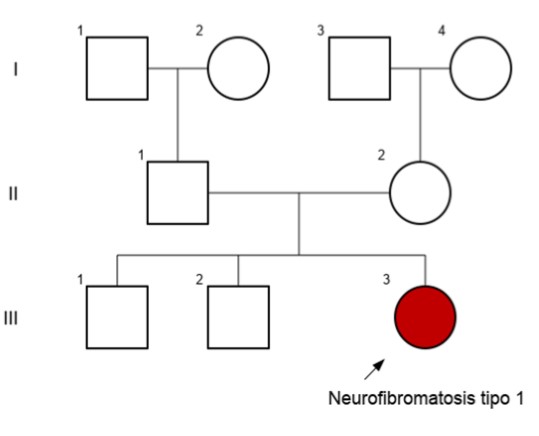
A los 4 meses de edad, la paciente presentó una masa de crecimiento progresivo en la región genital, de consistencia blanda y multilobulada, con compromiso del labio mayor izquierdo y ocupación del introito vaginal (Figura 1). El cuadro se acompañó de sangrado intermitente a nivel local. Además, se documentaron más de seis manchas café con leche mayores de 5 mm localizadas en el tronco y las extremidades. No se identificaron antecedentes patológicos familiares relevantes ni pertenencia a grupos étnicos o poblacionales minoritarios, siendo el único caso reportado en su familia (Figura 2).
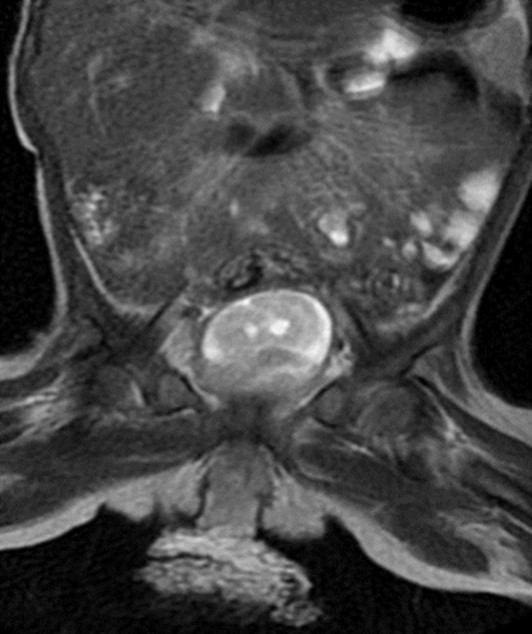
La resonancia magnética nuclear de abdomen y pelvis evidenció una lesión sólida de bordes definidos, con dimensiones de 3,9 × 3,4 × 6,5 cm, sin compromiso de órganos intraabdominales (Figura 3). La paciente fue llevada a resección quirúrgica de la masa, cuyo análisis histopatológico confirmó el diagnóstico de rabdomiosarcoma embrionario. Dada la presencia de una neoplasia maligna asociada a múltiples manchas café con leche, se realizó estudio molecular germinal mediante secuenciación de nueva generación, incluyendo análisis de deleciones y duplicaciones por algoritmos bioinformáticos. Se utilizó un panel dirigido a neurofibromatosis tipo 1 que incluyó los genes NF1, NF2 y SPRED1, identificándose una variante heterocigota en el gen NF1: c.730+2T>G (rs200962248), clasificada como patogénica según los criterios del Colegio Americano de Genética Médica y Genómica (ACMG) vigentes para 2025 (Richards et al., 2015). No se realizaron estudios de segregación en los padres debido a su negativa y ausencia de deseo de más descendientes.
En base a los hallazgos clínicos y moleculares, se estableció el diagnóstico de neurofibromatosis tipo 1 asociada a rabdomiosarcoma embrionario. La neoplasia fue clasificada como de bajo riesgo, grupo B, estadio T2M0N0, según el Estudio Intergrupal de Rabdomiosarcoma (IRS) de Estados Unidos. La tasa de supervivencia a cinco años para los casos de rabdomiosarcoma de bajo riesgo del grupo B puede superar el 80% y alcanzar hasta un 90% (NIH National Institutes of Health, 1988). El manejo oncológico consistió en poliquimioterapia adyuvante con vincristina, dactinomicina y ciclofosfamida. Dada la edad de la paciente y la localización anatómica del tumor, se optó por omitir radioterapia. Se ampliaron los estudios relacionados con la neurofibromatosis tipo 1 mediante resonancia magnética cerebral y de órbitas, sin encontrar hallazgos patológicos. Actualmente, la paciente se encuentra en seguimiento por oncología, genética y cirugía pediátrica, con adecuada evolución clínica y sin evidencia de recidiva tumoral ni alteraciones neurológicas en los estudios de imagen más recientes.
DISCUSIÓN
El mapeo óptico del genoma es una tecnología con gran aplicabilidad en la clínica, tanto en neoplasias como estudios constitucionales postnatales.
La neurofibromatosis tipo 1 es un síndrome genético de predisposición tumoral causado por la pérdida de función del gen NF1, el cual codifica la neurofibromina 1, una proteína que actúa como regulador negativo de la vía de señalización RAS/MAPK (Giugliano et al., 2019). Su inactivación conduce a una activación sostenida de RAS, lo que favorece la señalización proliferativa a través de receptores como el del factor de crecimiento epidérmico (EGFR), promoviendo la tumorogénesis (Mo et al., 2022).
El gen NF1 (OMIM*613113; 17q11.2) es uno de los más grandes del genoma humano, con 57 exones constitutivos y tres alternativos, y presenta múltiples pseudogenes, sin regiones comunes de mutación (“hot spots”). Esta complejidad estructural ha dificultado el análisis molecular y la interpretación funcional de muchas variantes (Mo et al., 2022). En la paciente que presentamos se identificó una variante germinal heterocigota en un sitio esencial de splicing GT-AG (c.730+2T>G, intrón 7), clasificada como patogénica según los criterios actualizados del ACMG para 2025 (Richards et al., 2015). Herramientas in silico como autoPVS1 predicen que esta alteración puede generar alteraciones graves en el procesamiento del ARN mensajero, como la omisión de exones o la aparición de nuevos sitios crípticos de splicing; la capacidad deletérea final suele estar provocada por la alteración del marco de lectura y la generación de codones de parada prematuros. Valero et al., 2011 reportaron la variante cercana c.730+1G>C en pacientes con diagnóstico confirmado de neurofibromatosis tipo 1, reforzando la importancia funcional de esta región (Valero et al., 2011).
En población pediátrica, se estima que el riesgo de desarrollar neoplasias malignas en individuos con neurofibromatosis tipo 1 se incrementa hasta en un 5 % respecto a la población general (McKeen et al., 1978). Entre las neoplasias más frecuentes se encuentran los gliomas de la vía óptica, neurofibrosarcomas, schwannomas malignos, leucemias mieloides, adenocarcinoma de la ampolla de Vater y rabdomiosarcomas (Huson y Hughes, 1993). Aunque el rabdomiosarcoma no es la neoplasia más común en esta condición, su asociación ha sido descrita en cerca de 100 casos reportados en la literatura médica (Margo, Johnson y Mancera, 2023).
Crucis et al., 2015, reportaron una serie de 16 pacientes (11 hombres y 5 mujeres) con esta asociación en un periodo de 20 años, en ocho centros franceses. En cuatro pacientes, el diagnóstico de neurofibromatosis tipo 1 se realizó de forma concomitante o posterior al diagnóstico del tumor, a partir de la identificación de manchas café con leche. La edad media al diagnóstico fue de 2,5 años (rango: 0,5–5,9 años), y en cinco casos el rabdomiosarcoma se diagnosticó durante el primer año de vida. La localización más frecuente fue pélvica (12 casos), y todos los tumores correspondieron al subtipo embrionario (Crucis et al., 2015). Por su parte, Castle et al., 2023, describieron una serie de pacientes pediátricos con diagnóstico inicial de rabdomiosarcoma que posteriormente fueron identificados con neurofibromatosis tipo 1. En los tres casos descritos, el diagnóstico tumoral se realizó antes del año de vida, todos los tumores fueron del subtipo embrionario y con localización pélvica o genitourinaria, sin enfermedad metastásica al momento del diagnóstico (Castle et al., 2023).
El análisis de la literatura sugiere que los casos de rabdomiosarcoma asociados a neurofibromatosis tipo 1 suelen diagnosticarse a edades más tempranas (antes de los 3 años en el 60 % de los casos), en comparación con los casos esporádicos, cuyo diagnóstico ocurre en promedio cerca de los 7 años. Asimismo, las localizaciones pélvicas o genitourinarias son más frecuentes en la asociación NF1–rabdomiosarcoma (64 %) que en los casos esporádicos (18–30 %), y el subtipo embrionario representa el 91 % de los casos asociados a neurofibromatosis tipo 1. En muchos de estos, el diagnóstico del rabdomiosarcoma precede la confirmación clínica de la neurofibromatosis tipo 1, lo que refuerza la importancia de un abordaje clínico-genético integral en pacientes pediátricos con neoplasias poco comunes y signos clínicos sutiles de RASopatías (Margo, Johnson y Mancera, 2023; Castle et al., 2023; Crucis et al., 2015).
El manejo de pacientes con neurofibromatosis tipo 1 requiere un abordaje integral y multidisciplinario, incluyendo pediatras o internistas, neurólogos, oftalmólogos, ortopedistas, genetistas y neurocirujanos, según las manifestaciones clínicas. Ante la presencia de una neoplasia maligna, estos pacientes representan un subgrupo con características particulares, dado su riesgo aumentado de efectos adversos a la quimioterapia y mayor susceptibilidad al desarrollo de tumores secundarios inducidos por radiación (Castle et al., 2023). En este contexto, un enfoque terapéutico más conservador y una vigilancia activa podrían ser apropiados. Además, se están evaluando terapias dirigidas contra la vía RAS/MAPK, como inhibidores de MEK (ej. selumetinib) o mTOR (ej. sirolimus), especialmente en casos refractarios o recurrentes, aunque aún no forman parte del tratamiento estándar (Crucis et al., 2015).
Este caso resalta la importancia de considerar síndromes de predisposición genética como neurofibromatosis tipo 1 ante el diagnóstico de rabdomiosarcoma en edades atípicas o con localización genitourinaria, especialmente cuando coexisten signos clínicos sugerentes como manchas cafés con leche. La identificación oportuna de estas asociaciones tiene implicaciones pronósticas, terapéuticas y de seguimiento a largo plazo, tanto para el paciente como para su entorno familiar.
Declaración de conflicto de intereses
Los autores declaran que no existe ningún conflicto potencial de intereses.
Los autores no recibieron apoyo económico de ninguna organización para la elaboración de este manuscrito.
Aspectos éticos
Se tuvo en cuenta las consideraciones éticas y precauciones para garantizar la confidencialidad del paciente, se firmó consentimiento informado por parte de la madre del paciente para la toma de fotografías y publicación del caso.


