
En los últimos siete años, la secuenciación del transcriptoma o ARN de un individuo (RNA-seq) ha emergido de manera constante como un ensayo complementario a la ya bien establecida secuenciación del ADN para el diagnóstico y descubrimiento de enfermedades raras. La RNA-seq ha sustituido principalmente a los métodos dirigidos a regiones genómicas específicas (Mortazavi et al., 2008; Nonis et al., 2014; Stark et al., 2019), debido a que es capaz de analizar todos los genes expresados (entre diez mil y doce mil, de media, dependiendo principalmente del tejido).
En el diagnóstico de enfermedades raras, la RNA-seq se ha utilizado principalmente para i) confirmar el efecto de variantes patogénicas candidatas obtenidas a partir del ADN que sirven como «estudio funcional bien establecido que muestra un efecto deletéreo» siguiendo las directrices del ACMG (American College of Medical Genetics and Genomics) (Richards et al., 2015) e ii) identificar genes candidatos en los que inicialmente no se encontraron variantes o no se priorizaron. En este artículo de Comentario, describo algunos de los estudios que han utilizado con éxito RNA-seq para el diagnóstico de enfermedades raras, los diferentes métodos estadísticos especializados que han surgido, así como las limitaciones de la tecnología y las perspectivas de futuro.
ESTUDIOS
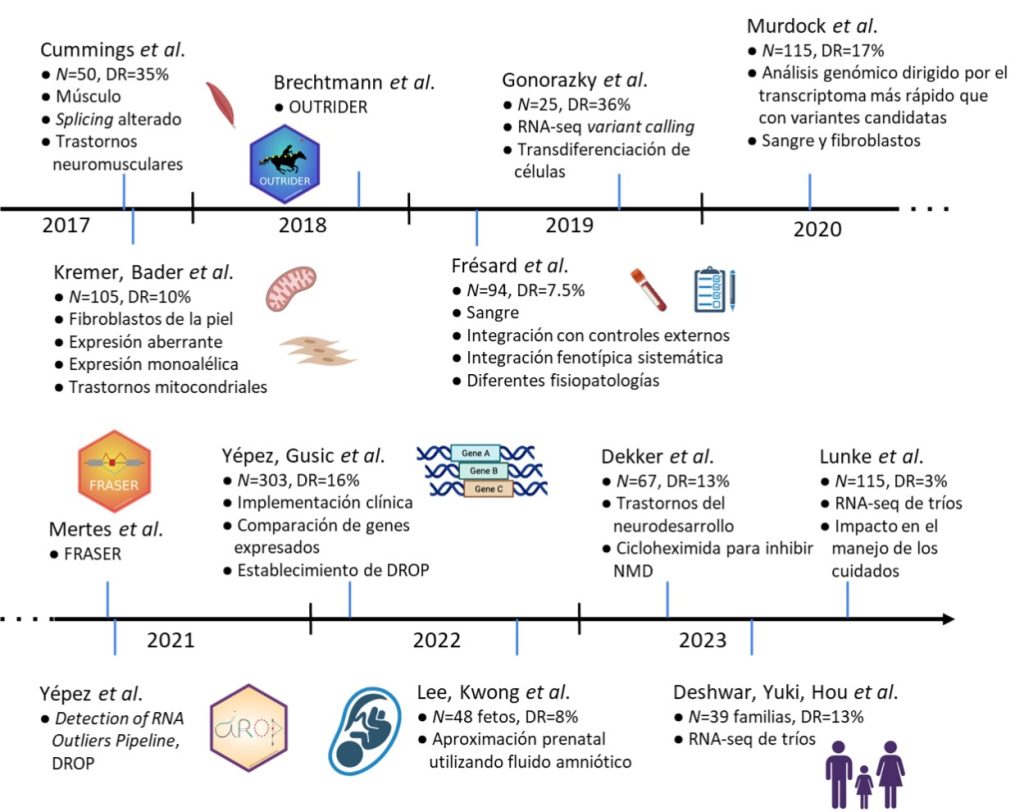
Los estudios pioneros de Kremer y colaboradores de la Universidad Técnica de Múnich y de Cummings y colaboradores del Instituto Broad, lanzados simultánea e independientemente en 2016 y publicados en 2017, originaron el concepto y la posibilidad de utilizar RNA-seq para el diagnóstico de enfermedades raras. La idea era la misma: reunir una cohorte suficientemente grande y analizar todos los genes expresados en una muestra afectada para detectar desviaciones significativas en la expresión y el procesado del ARN con respecto a la media de la cohorte. A continuación, encontrar variantes genéticas que pudieran explicar esas alteraciones y, por último, evaluar si podían ser la causa de la enfermedad en combinación con el fenotipo del paciente (Fig. 1).
El primer estudio secuenció fibroblastos derivados de la piel y el segundo, músculo, estableciendo así la utilidad de esos tejidos para esta tarea. Un par de años después, Frésard y colaboradores de Stanford publicaron un estudio similar con sangre. Esos tres tejidos clínicamente accesibles se han convertido en los más utilizados, cada uno con sus pros y sus contras. Por ejemplo, el músculo es sin duda el estándar para estudiar los trastornos (neuro)musculares, ya que la mayoría de los genes de interés sólo se expresan en él. La sangre es el tejido más accesible; sin embargo, tiene el menor número de genes expresados en comparación con los otros dos (Yépez et al., 2022). Los fibroblastos parecen ser el tejido más completo, ya que se expresan la mayoría de los genes relacionados con enfermedad y es posible cultivar las células y reutilizarlas para otros ensayos.
Desde entonces se han publicado muchos otros estudios que informan de una variedad de trastornos: mitocondriales, neuromusculares, neurológicos, del neurodesarrollo e inmunológicos, procedentes de diferentes centros de todo el mundo, como Canadá (Gonorazky et al., 2019; Deshwar et al., 2023), Estados Unidos (Murdock et al., 2021), Alemania (Yépez et al., 2022), Hong Kong (Lee et al., 2022), Países Bajos (Dekker et al., 2023) y Australia (Lunke et al., 2023). El aumento registrado en los rendimientos diagnósticos oscila entre el 7% y el 36%, dependiendo de la enfermedad, la metodología y el tejido explorado, pero especialmente de si la cohorte incluía pacientes con variantes candidatas o no (Fig. 2). Muy recientemente, Deshwar y colaboradores, del Hospital for Sick Children, así como Lunke y colaboradores de múltiples centros de Australia, exploraron la utilidad de realizar RNA-seq en trío, como se hace frecuentemente en los diagnósticos basados en ADN. No se observó un beneficio diagnóstico significativo del diseño en trío con respecto a la realización de pruebas únicamente en el caso índice. Sin embargo, dado que en el estudio participó un número modesto de familias, es necesario realizar más investigaciones para concluir su utilidad.
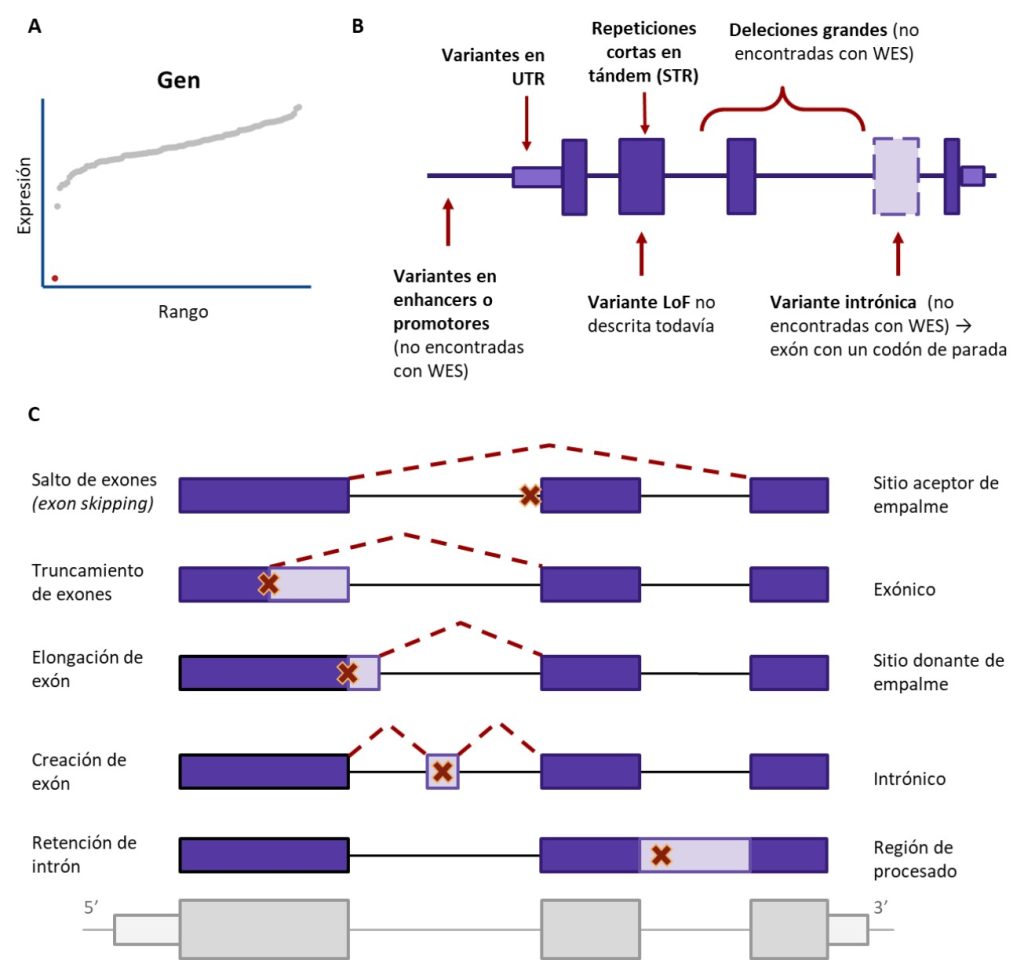
MÉTODOS ESPECIALIZADOS
Tras el éxito de los dos estudios pioneros, se empezaron a desarrollar métodos especializados para detectar la expresión aberrante y el procesado alternativo o splicing. Los métodos modelan los recuentos de lecturas con una distribución estadística (generalmente binomial negativa para recuentos de lecturas y beta-binomial o Dirichlet-multinomial para recuentos de empalmes) y luego obtienen valores p para cada combinación gen-muestra o empalme-muestra. Algunos de estos métodos son OUTRIDER (Brechtmann et al., 2018) y OutSingle (Salkovic et al., 2023) para la expresión; y Brisbee (Halperin et al., 2021), FRASER (Mertes et al., 2021), FRASER2 (Scheller et al., 2023), y LeafCutterMD (Jenkinson et al., 2020) para el empalme. OUTRIDER y FRASER2 están incluidos en el Detection of RNA Outliers Pipeline, DROP (Yépez et al., 2021), una solución integral para facilitar y acelerar la detección de valores atípicos a partir de datos brutos.
Estos métodos pudieron probarse a fondo aprovechando el amplio recurso proporcionado por el proyecto The Genotype-Tissue Expression (GTEx), que ofrece más de 8000 muestras emparejadas WGS-RNA-seq de casi 1000 individuos y 51 tejidos (GTEx Consortium, 2017). Con este recurso, fue posible establecer que los valores atípicos de expresión y empalme, efectivamente, están asociados con variantes raras (Zeng et al., 2015; Li et al., 2017) y, por lo tanto, pudo convertirse en un conjunto de datos de referencia para los métodos de detección de valores atípicos.
LIMITACIONES
La extracción de ARN se limita a tejidos accesibles, como la sangre y la piel y, en casos extremos, el músculo. Esto implica que el estudio se restringe a los genes expresados en el tejido investigado: alrededor del 65% de los genes causales de enfermedades mendelianas conocidas (OMIM) se expresan en la sangre y el 70% en los fibroblastos derivados de la piel. El año pasado, un estudio pionero de la Universidad de Hong Kong demostró que la RNA-seq también puede hacerse a partir del líquido amniótico durante el embarazo para evaluar la expresión y el procesado del ARN del feto (Lee et al., 2022). Por último, un reciente artículo preprint ha descrito la posibilidad de analizar el ARN de otros tejidos accesibles: hisopos bucales, folículos pilosos, saliva y pellets de células de la orina (Martorella et al., 2023). Es necesario investigar a fondo para verificar su utilidad en el diagnóstico.
Otra limitación importante de la RNA-seq es que el efecto de la abundante clase de variantes con cambio de sentido podría no reflejarse en el transcriptoma. La proteómica tiene la ventaja de captar el efecto de las variantes con cambio de sentido y los cambios reguladores postranscripcionales; sin embargo, carece de la potencia necesaria para revelar el procesado del ARN erróneo y la expresión alelo-específica. Por ahora, la RNA-seq y la proteómica parecen más complementarias que competidoras (Kopajtich et al., 2021; Vialle et al., 2022).
Desafíos públicos
Observando los beneficios potenciales de la tecnología, se han lanzado desafíos públicos para proporcionar diagnósticos genéticos utilizando ADN, ARN y datos clínicos a partir de una cohorte de enfermedades raras, para que grupos de todo el mundo puedan utilizar sus herramientas y enfoques. Ejemplos de ello son el reto CAGI 6 (Critical Assessment of Genome Interpretation)1 «Predecir eventos moleculares subyacentes a la enfermedad a partir del genoma y transcriptoma de un paciente» ofrecido por el Hospital for Sick Children‘s y el reto Kaggle EndALS (Esclerosis Lateral Amiotrófica)2, ambos lanzados en 2021. Además, eventos tipo Hackathon in situ, como el International Undiagnosed Hackathon3 organizado por el UDNI en el Instituto Karolinska en 2023, ofrecen RNA-seq y WGS acoplados para acelerar el diagnóstico. La creciente popularidad de esta tecnología, especialmente en combinación con el análisis de ADN, es innegable.
1 https://genomeinterpretation.org/cagi6-sickkids.html
2 https://www.kaggle.com/datasets/alsgroup/end-als
3 https://www.undiagnosedhackathon.org/
LO QUE NOS DEPARA EL FUTURO
La secuenciación de ARN forma parte de la denominada Secuenciación de Próxima Generación. Poco a poco, está pasando a llamarse RNA-seq de lectura corta, ya que está surgiendo una nueva tecnología que permite secuenciar lecturas mucho más largas, la RNA-seq de lectura larga (Pollard et al., 2018; Wang et al., 2023). Su potencial y su mejora en el diagnóstico de enfermedades raras aún están por demostrarse. Además, la secuenciación de genoma completo ahora permite detectar variaciones en el número de copias (CNV) en forma de grandes deleciones, duplicaciones, inserciones, inversiones, translocaciones y expansiones de repeticiones. Cómo puede la RNA-seq detectar cada una de ellas empezó a explorarse en adultos de edad avanzada e individuos con enfermedad de Alzheimer en el minucioso estudio de (Vialle et al., 2022), pero es necesario seguir investigando.
Obtener un diagnóstico es útil, ya que puede conducir a proporcionar asesoramiento genético. Sin embargo, el objetivo principal es proporcionar tratamiento, por ejemplo, mediante la administración de suplementos farmacológicos (Koch et al., 2017). La RNA-seq puede ser especialmente útil para orientar oligonucleótidos antisentido, como han hecho recientemente Kumar y sus colegas, que diseñaron y aplicaron estos oligonucleótidos antisentido para restaurar el procesamiento normal del ARNm TIMMDC1 y los niveles de proteína en las células de los pacientes (Kumar et al., 2022).
Por último, el mayor cambio podría producirse cuando las compañías de seguros reembolsen las pruebas de ARN, como ya está empezando a ocurrir con el ADN.
Declaración de conflicto de intereses
El autor declara la ausencia de conflicto de intereses.


