INTRODUCCIÓN
La displasia atriodigital, también conocida como síndrome corazón-mano, se refiere a un grupo de trastornos congénitos que se caracterizan por malformaciones de las extremidades superiores y el corazón. El Síndrome de Holt-Oram, en adelante SHO (OMIM 142900/ORPHA392), es una de las formas más frecuentes y mejor conocidas de estos síndromes (Zaragoza, 2016). Fue descrito por Mary Clayton Holt y Samuel Oram en 1960 (Holt-Oram, 1960) y se caracteriza por una disfunción del desarrollo embrionario con malformaciones en las extremidades superiores, implicando a huesos radiales, tenares o hipotenares (Iwanicka-Pronicka, 2016), junto con un historial familiar de malformación cardiaca congénita, principalmente defecto atrial septal de tipo ostium secundum (Su W, 2016). Se han descrito anomalías en el sistema de conducción, como la fibrilación auricular paroxística (Guo, 2016), a veces asociada con trastornos de la frecuencia cardiaca así como varios grados de bloqueo auriculoventricular. Rara vez asocia otro tipo de malformaciones, como las orales y maxilofaciales (Arslanoglu, 2016) entre otras.
El SHO es una enfermedad ultra rara, se estima una prevalencia de 1 por cada 100.000 nacidos vivos (Ali, 2016) y está causado por mutaciones en el gen TBX5, localizado en 1966 en el cromosoma 12 (12q24.1). El gen TBX5 codifica el factor de transcripción T-box5 (D’Aurizio 2016) que regula la cardiogénesis, mediante la expresión de otros genes implicados en el desarrollo cardíaco y de las extremidades durante la embriogénesis. Hasta la fecha se han encontrado pacientes con SHO con casi un centenar de mutaciones del gen TBX5, la mayoría de los cuales causan un truncamiento prematuro de la transcripción primaria produciendo haploinsuficiencia (Steimle, 2016), una situación en la cual la proteína producida por una sola copia de un gen normal no es suficiente para garantizar una función normal.
Se ha de tener en cuenta que la mayoría (90%) de las alteraciones genéticas que producen cambios en el gen TBX5 son producidas de novo pero, una vez producida la alteración, el SHO se hereda de forma autosómica dominante, y tiene una elevada penetrancia con una expresividad variable (Barisic, 2014). Más del 85% de los individuos diagnosticados clínicamente son portadores de una mutación TBX5. Sin embargo en el resto de casos desconocemos la causa, existiendo algunas publicaciones que hablan de la duplicación de algunos exones del TBX5 (Kimura, 2015).
PACIENTES Y MÉTODOS
Se realiza interconsulta desde Cardiología a la Unidad de Genética Médica de nuestro hospital de una paciente de 33 años, con una malformación cardiaca conocida y que ha comenzado con trastornos del ritmo. Se trata de una mujer adulta, operada en la infancia de una comunicación interauricular tipo seno venoso, que asocia una hipoplasia de la eminencia tenar y que, en la actualidad, padece de una fibrilación auricular persistente con PR corto.
Al realizar el genograma (ver figura 1) se evidencia una posible herencia autosómica dominante de una malformación cardiaca congénita. La madre de la paciente toma antihipertensivos y presenta una aurícula izquierda levemente dilatada, sin otra patología asociada. La única hermana de la paciente falleció de muerte súbita a los 27 años por una cardiopatía no aclarada y la única hija de la paciente, de 9 años, está intervenida de una comunicación interauricular tipo seno venoso también. Valoramos estudiar el gen TBX5 entre otros y se cita a la familia a revisión.
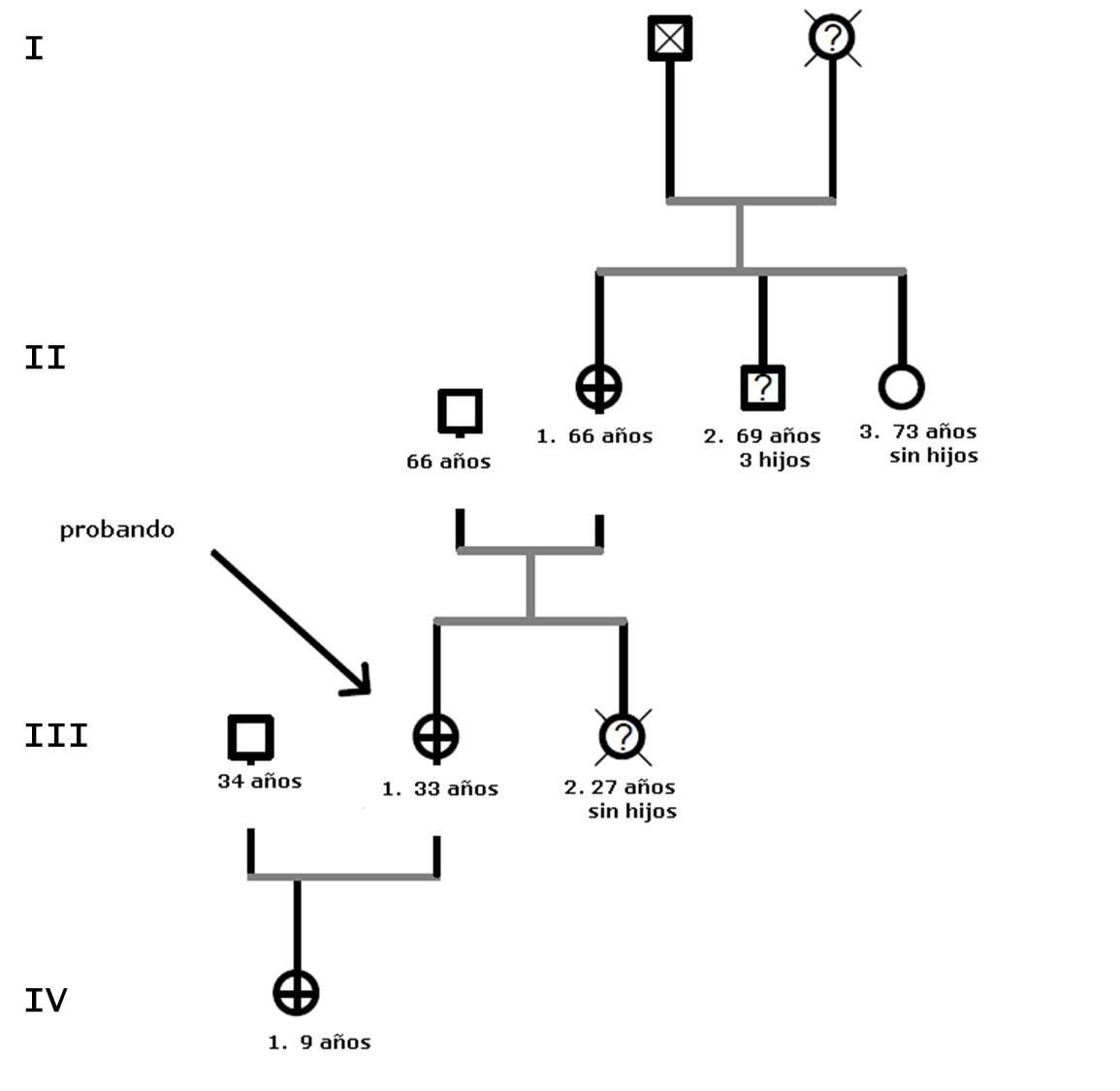
Al cabo de un mes recibimos en consulta a la paciente, a la madre y a la hija de la primera. La abuela solo objetiva hipertensión arterial pero la nieta presenta ambos antebrazos cortos, por lo que se cursa interconsulta urgente a Traumatología Infantil de nuestro hospital. Se le plantea estudio genético y, una vez obtenido el correspondiente consentimiento informado, la metodología empleada en nuestro laboratorio externo fue la siguiente:
- Extracción del ADN utilizando el kit comercial QIAamp DNA Blood Mini Kit (Qiagen).
- Cuantificación del ADN obtenido mediante la visualización del mismo en geles de agarosa al 0.8%.
- Análisis de deleciones/duplicaciones de los genes relacionados con la enfermedad congénita cardiaca: hibridación con sondas específicas para cada uno de los exones que contienen los genes GATA4, NKX2-5, TBX5, BMP4 y CRELD1 y posterior amplificación por PCR de las regiones hibridadas, mediante la técnica MLPA (Multiple Ligationdependent Probe Amplication), con marcaje fluorescente y detección mediante electroforesis capilar en condiciones desnaturalizantes en un ABI PRISM 310 Genetic Analyzer, de los fragmentos ligados y amplificados.
- Secuenciación completa del gen TBX5: para la amplificación del gen se empleó el sistema VariantSEQr de Applied Biosystems utilizando un set de primers específicos para la amplificación por PCR y posterior secuenciación de las regiones codificantes y regiones intrónicas adyacentes del gen TBX5. Una vez purificado el producto de PCR mediante la enzima exonuclease ExoSAP-IT, éste fue secuenciado usando el kit comercial BigDye 3.1 de Applied Biosystems y posterior purificación de la secuenciación mediante columnas de purificación.
- El proceso de electroforesis capilar se llevó a cabo en un analizador genético ABI 3130 y las secuencias obtenidas fueron analizadas con un software específico: SeqScape de Applied Biosystems.
RESULTADOS
La muestra analizada de la paciente no presentaba deleciones ni duplicaciones en ninguno de los exones de los diferentes genes analizados con el ensayo de MLPA (GATA4, NKX2-5, BMP4 y CRELD1). El cuadro clínico de la paciente no era debido pues a deleciones ni duplicaciones en los estos cuatro genes.
Tampoco el cuadro clínico de la paciente era debido a la presencia de mutaciones puntuales en ninguno de los exones codificantes analizados, ni en las regiones intrónicas adyacentes estudiadas, para el gen TBX5. Sin embargo la muestra analizada si presentaba una duplicación para todos los exones del gen TBX5. Este resultado puede ser compatible con el cuadro clínico de la paciente y causante del SHO.
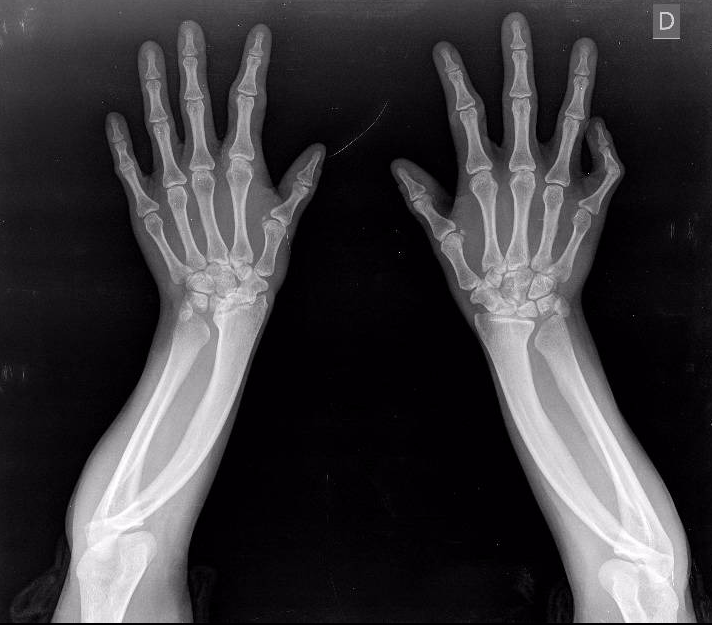
Una vez estudiada a su hija en la Consulta de Traumatología Infantil se evidencia luxación congénita de cabeza radial bilateral, mano zamba cubital bilateral por acortamiento de cúbito, e hipoplasia a nivel de pulgar bilateral pero con eminencia tenar útil (figura 2) que apoyan el diagnóstico del SHO atípico también en la hija. Es importante resaltar que nos encontramos ante un síndrome de Holt Oram atípico, puesto que en este síndrome las manifestaciones esqueléticas son tipícamente radiales y en menor grado y menos intensas en el lado cubital. En la paciente, como se ve en las RX hay una angulación del radio, con un acortamiento de cúbito que recuerda mucho la deformidad de Madelung, mientras que las anomalías del lado radial es fundamentalmente un acortamiento del 1º metacarpiano. Ante estos datos se decide estudio familiar del TBX5 en abuela y nieta, con idéntico resultado. No así el estudio traumatológico al no disponer de imágenes radiológicas de la madre de la paciente.
DISCUSIÓN
Las mutaciones en el gen TBX5 son la principal causa (85%) del síndrome atriodigital descrito por Holt y Oram. Es un gen que se compone de 10 exones y está situado en el brazo largo del cromosoma 12 (12q24.1) (Nourzad, 2011). El TXB5 es un miembro de una familia de factores de transcripción T-box que codifican los factores de transcripción implicados en el desarrollo celular embrionario, la regulación del desarrollo de estructuras extraembrionarias, así como en muchos aspectos de la organogénesis (Dreßen, 2016). Dentro de la familia T-box la expresión del TBX5 es muy importante para la iniciación de la extremidad superior y el desarrollo del tabique y el sistema eléctrico cardíaco, no existiendo antecedentes de malformaciones en la extremidad inferior.
Pero la relación entre las alteraciones del gen TBX5 y la aparición del SHO es variable (Sinha, 2015). De este modo hay estudios que hablan de un tercio de los pacientes con fenotipo de SHO que asocien dichas alteraciones, por lo que el estudio únicamente del gen TBX5 en estos casos sería insuficiente. Sin embargo esta proporción aumenta significativamente en caso de hacer un diagnóstico exhaustivo de los signos del síndrome, de este modo la consulta de genética se hace casi imprescindible a la hora de aglutinar todos estos signos en un síndrome heredofamiliar.
En este caso la variación en las alteraciones del gen es amplio siendo, la mayoría de estas, mutaciones que inhabilitan la funcionalidad de la proteína TBX5. Se han encontrado pacientes con SHO con más de 90 mutaciones del gen TBX5 (Basson, 1999), la mayoría de los cuales causan un truncamiento prematuro de la transcripción TBX5 primaria produciendo haploinsuficiencia (Steimle, 2016), una situación en la cual la proteína producida por una sola copia de un gen normal no es suficiente para garantizar una función normal. Por otra parte un aumento de la dosificación del gen TBX5, tal y como se produciría en una duplicación del cromosoma 12, de la región que contiene el gen, también daría lugar a anomalías cardíacas, por lo que la sobreexpresión de TBX5 afectaría a la cardiogénesis (Kim, 2016). Por el contrario una pérdida de la dosificación del gen, como por ejemplo en una deleción del cromosoma 12, de la región que contiene el gen, volvería a dar lugar a anomalías cardíacas al provocar una expresión incompleta de la proteína TBX5 (Shamseldin, 2016).
Diversos estudios hablan de deleciones del gen TBX5 en familias con SHO pero sin mutaciones que lo expliquen, sin embargo muy pocos estudios hablan de duplicaciones en este gen. La duplicación de TBX5 ha sido identificada en una familia japonesa (Kimura, 2015) y otra británica (Patel 2012); sin embargo, el genotipo de la primera familia fue incompleto y el fenotipo de la segunda, además, fue atípico. En nuestro estudio, que abarca una duplicación de todos los exones, se confirmó en una persona con el típico fenotipo del SHO. La madre de la paciente no presentaba características clínicas destacables y la paciente sólo presentaba una cardiopatía congénita intervenida, con una fibrilación auricular (Wang, 2016), junto a una pequeña hipoplasia tenar. Es la hija de la paciente la única que presenta un fenotipo clínico característico de SHO, cardiaco (Naik, 2016) y de miembros superiores, mientras que las tres presentan la duplicación de todos los exones del TBX5. Esto nos hace pensar que, además de una variabilidad evidente (Barisic, 2014), también existe una suerte de sobreexpresión con cada generación. Esta variabilidad intrafamiliar tan amplia, unido al aumento de los signos en cada generación, nos lleva a la conclusión de que debamos ampliar estudios.
Si el gen TBX5 está formado por 10 exones no todos ellos tienen la misma importancia. Mientras que los exones 3 a 10 son la región codificante del gen los dos primeros exones no son traducidos. Sin embargo, para la normal expresividad de dicho gen, todos los exones son relevantes. Muchos estudios abundan en la importancia de los exones 1 y 2, y su funcionamiento normal, para la aparición del síndrome (Patel, 2012) aunque, a priori, no sean traducidos. Creemos que nuestro estudio es el único que habla de una duplicación total del TBX5 que causa un SHO en un miembro de una familia mientras que, en un familiar de primer grado, no existe suficiente información. Se necesitan más estudios funcionales para establecer los efectos de la duplicación y el mecanismo patogénico pensando, incluso, en la epigenética.
Para avanzar en la comprensión de la correlación fenotipo-genotipo entre las mutaciones TBX5 y el SHO hacemos hincapié en que, además de las mutaciones del gen TBX5 implicadas (Heinritz, 2005), la detección de duplicaciones debe ser una parte rutinaria del análisis molecular de los pacientes. De este modo, si tras una exhaustiva exploración física del paciente sospechamos de un SHO, y este no presenta mutación alguna, deberemos ampliar estudios mediante la técnica MLPA, buscando deleciones o duplicaciones del gen.
CONCLUSIONES
El síndrome de Holt-Oram, presenta un patrón de herencia autosómico dominante (Rodagi, 2016). Estos resultados tienen implicaciones hereditarias y familiares. Sería recomendable poder estudiar la presencia o no de esta mutación en los familiares directos así como en los hermanos de la progenitora de la paciente. Así mismo es importante reseñar que faltan estudios radiológicos de imagen a la madre de la paciente.
Conviene recordar que los descendientes de la paciente tienen el 50% de probabilidad de contraer dicha alteración genética. Se ha de tener en cuenta que la mayoría de las alteraciones genéticas que producen cambios en el gen TBX5 son producidas de novo (Dreßen, 2016), por tanto sería normal que los progenitores de la madre de la paciente no hubiesen presentado esta alteración. En cualquier caso es importante realizar un estudio a los tíos maternos de la paciente para descartar segregación familiar.
Todo esto no excluye que la paciente pueda transmitir con un 50% de probabilidad esta alteración a sus futuros descendientes (Rodagi, 2016), por lo que necesario dar un adecuado asesoramiento genético a la paciente y a su hija cuando esta alcance su mayoría de edad o inicio de la edad reproductiva.
CONFLICTO DE INTERESES
Declaramos la ausencia total de conflictos de intereses en este trabajo.

