INTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) se caracteriza por un aumento del grosor de la pared ventricular que no se explica por las condiciones de carga. Se trata principalmente de un trastorno autosómico dominante causado por variantes genéticas que afectan a las proteínas del sarcómero, esenciales para la función contráctil del corazón. El gen MYBPC3 es uno de los más comúnmente implicados. Concretamente, las variantes patógenas de MYBPC3 representan entre el 40 % y el 45 % de los casos de MCH, y todos los genes descritos explican entre el 80 % y el 85 % de los casos (Chiswell, Zaininger y Semsarian, 2023).
La MCH es la miocardiopatía hereditaria más común, con una prevalencia estimada del 0,2-0,5 % en todo el mundo, que afecta al menos a 1 de cada 500 personas. Aunque suele ser autosómica dominante (con un 50 % de probabilidades de herencia), también existen formas autosómicas recesivas, que son más comunes en familias consanguíneas. La mayoría de las variantes causantes son de tipo missense (cambio de aminoácido), pero también hay variantes de desplazamiento del marco de lectura o truncamiento que pueden alterar significativamente la proteína (Chiswell, Zaininger y Semsarian, 2023).
La proteína cMyBP-C, codificada por MYBPC3, es una proteína estructural vital que mantiene la integridad del sarcómero y regula la interacción actina-miosina, la cinética de contracción y la sensibilidad a los iones de calcio.
Aproximadamente el 50% de las variantes en MYBPC3 dan lugar a productos proteicos truncados, lo que a menudo conduce a una haploinsuficiencia a través de mecanismos como la degradación del ARN mensajero mediada por mutación terminadora (NMD en sus siglas en inglés) y el sistema ubiquitina-proteasoma. Algunas proteínas mutantes pueden actuar como “péptidos tóxicos”. Las variantes no truncadas (variantes sin sentido y pequeñas inserciones/deleciones en el marco de lectura) representan alrededor del 15 % de los casos de MCH relacionados con MYBPC3. Aunque sus mecanismos son menos conocidos, también pueden causar haploinsuficiencia o afectar a la función/estabilidad de la proteína. (Tudurachi et al., 2023)
La amplia variabilidad fenotípica de la MCH causada por variantes en MYBPC3 puede abarcar desde casos leves hasta graves, con un mayor riesgo de muerte súbita cardíaca. Aunque generalmente se asocian con una aparición más tardía y un fenotipo menos grave que las variantes en MYH7, algunos estudios sugieren que pueden causar disfunción ventricular y susceptibilidad a arritmias. (Tudurachi et al., 2023)
Los portadores de variantes doble heterocigotas, heterocigotas compuestas y homocigotas suelen presentar formas más graves de miocardiopatía que pueden provocar una muerte prematura. Las causas no sarcoméricas representan hasta el 35 % de los casos de MCH en niños (enfermedades neuromusculares, enfermedades mitocondriales, errores congénitos del metabolismo). Además, la MCH con un inicio antes del primer año de vida tiene un pronóstico más desfavorable (Melas et al., 2023)
Las directrices actuales recomiendan encarecidamente la realización de pruebas genéticas utilizando paneles específicos que emplean secuenciación de nueva generación (NGS) para todos los pacientes con MCH, con el fin de confirmar el diagnóstico, identificar la etiología molecular y orientar el cribado y el tratamiento. La tasa de éxito diagnóstico de las pruebas genéticas es de aproximadamente el 30 % en los casos esporádicos y de hasta el 60 % en los casos familiares o en pacientes más jóvenes con hipertrofia septal asimétrica típica. La interpretación de los resultados y la clasificación de las variantes son los aspectos más difíciles y críticos de las pruebas genéticas, debido a la complejidad de la variación genética humana y a los datos disponibles, que son limitados o ambiguos. (Abbas et al., 2024)
En las últimas décadas, la genómica se ha convertido en una herramienta esencial para comprender la base molecular de las enfermedades cardiovasculares hereditarias. El desarrollo de tecnologías de secuenciación masiva ha permitido identificar variantes genéticas causales en genes clave relacionados con las enfermedades cardíacas, muchas de las cuales tienen implicaciones directas para el pronóstico y el manejo clínico. Así, genes como MYBPC3, MYH7 y PLN, cuyas variantes patógenas se asocian con miocardiopatías con alto riesgo de muerte súbita o insuficiencia cardíaca precoz.
La evidencia acumulada ha sido tan sólida que diversas organizaciones internacionales, como la ACMG/AMP, han recomendado la notificación sistemática de las variantes patógenas y probablemente patógenas en estos genes, incluso cuando se identifican como hallazgos secundarios en estudios genómicos indicados por otros motivos. Todo ello se debe a la elevada morbilidad y mortalidad asociadas a estas afecciones, así como a la existencia de estrategias terapéuticas y de seguimiento clínico que pueden prevenir complicaciones graves. Sin embargo, la interpretación de las variantes de VUS sigue planteando un reto, especialmente en pediatría, donde la integración temprana de la genética en el diagnóstico diferencial puede ser decisiva para un enfoque clínico oportuno y preciso (Lee et al. 2025).
Los avances en los estudios genómicos a gran escala han permitido redefinir la arquitectura genética de las cardiopatías congénitas, demostrando que un número significativo de casos se debe a variantes dominantes, tanto de novo como heredadas. El análisis de una cohorte de más de 11 000 pacientes identificó un exceso significativo de variantes heterocigotas perjudiciales en al menos 60 genes con implicaciones directas para el desarrollo cardíaco. Estas variantes representaban aproximadamente el 10% de los casos, con una expresión fenotípica variable que incluía tanto defectos cardíacos aislados como síndromes con afectación extracardíaca o del desarrollo neurológico. Estos hallazgos respaldan el papel fundamental de la genómica como herramienta de diagnóstico integral para detectar complicaciones sistémicas y adaptar el seguimiento clínico personalizado en pacientes con cardiopatías congénitas. (Sierant et al., 2025).
Se ha identificado una contribución significativa de las variantes recesivas a la etiología de las cardiopatías congénitas, especialmente en poblaciones con un alto grado de consanguinidad. En un análisis sistemático de más de 5000 casos, la estimación de variantes recesivas representa al menos el 2,2 % de los casos, con una distribución desigual según el tipo de malformación y los antecedentes genéticos. En particular, el análisis mostró una fuerte asociación entre las variantes bialélicas en genes, concretamente MYH6, GDF1 y MYBPC3, y los fenotipos del lado izquierdo del corazón, incluida la obstrucción del tracto de salida del ventrículo izquierdo, lo que sugiere que la disfunción contráctil durante las primeras etapas del desarrollo puede ser un mecanismo patogénico subyacente. Estos hallazgos subrayan la importancia de tener en cuenta los patrones de herencia recesiva en la evaluación genómica de los pacientes con cardiopatías complejas, incluso en ausencia de antecedentes familiares claros (Dong et al., 2025).
Este caso clínico destaca la importancia de utilizar la genómica y otras herramientas multimodales, incluido el fenotipado reverso, en favor de la medicina personalizada, con el fin de integrar todos los recursos disponibles actualmente y lograr una correlación genotipo-endotipo-fenotipo y obtener un diagnóstico específico, evitando así perder oportunidades para la estratificación del riesgo, la vigilancia clínica estricta, los tratamientos personalizados, el seguimiento, el pronóstico y la medicina anticipatoria-preventiva, junto con la importancia del asesoramiento sobre el riesgo genético familiar.
DESCRIPCIÓN DEL CASO
Paciente varón, de 1 año y 8 meses de edad, nacido a término del primer embarazo, con buen peso y altura al nacer. Los padres no son consanguíneos y ambos son asintomáticos. No se dispone de pruebas genéticas para ellos. El embarazo tuvo un inicio tardío de la atención prenatal y la madre tenía antecedentes de síndrome de ovario poliquístico, hiperprolactinemia, infecciones recurrentes del tracto urinario inferior que requieren antibióticos de amplio espectro y uso de cabergolina y antecedentes perinatales de amenaza de aborto espontáneo y parto prematuro. No hubo anomalías en la ecografía prenatal. El paciente tenía antecedentes familiares maternos de abuela y bisabuela diagnosticadas con arritmia no especificada, sin otros familiares con antecedentes
de enfermedades cardiovasculares genéticas congénitas identificadas. A los 5 meses de edad, el paciente requiere una cirugía a corazón abierto para cerrar una comunicación interventricular (VSD), un foramen oval permeable (PFO), un defecto del tabique auricular (AICD), un conducto arterioso permeable (PDA) y la reconstrucción de la arteria pulmonar con pericardio autólogo.
En la exploración física, el paciente presenta bajo peso y estatura para su edad, con una ligera hiperlaxitud ligamentosa y un desarrollo neurológico adecuado sin otras anomalías fenotípicas. Se encuentra en seguimiento multidisciplinar por parte de neurología debido a un único episodio ictal de causa desconocida en la infancia, sin lesiones anatómicas ni foco epiléptico, sin tratamiento farmacológico, solo bajo vigilancia clínica. Dado su bajo peso y estatura para su edad, se encuentra en seguimiento con gastroenterología con recomendaciones dietéticas, y se ha descartado que padezca un trastorno metabólico hereditario. Debido al asma leve persistente controlada y al síndrome de apnea obstructiva del sueño-hipopnea (SAOS) leve documentado en la polisomnografía, se encuentra en seguimiento por neumología pediátrica y se le trata con esteroides inhalados.
El último informe ecocardiográfico muestra el estado postoperatorio tardío del cierre quirúrgico de la comunicación interauricular, la comunicación interventricular y el conducto arterioso persistente, sin evidencia de derivación residual, geometría ventricular izquierda conservada con función sistólica normal (FEVI: fracción de eyección ventricular izquierda del 61 %), diástole normal, ventrículo derecho con morfología normal y rendimiento sistólico-diastólico adecuado, sin evidencia ecocardiográfica de hipertensión arterial pulmonar. El electrocardiograma muestra un ritmo sinusal con trastornos leves de repolarización intrínseca.
Teniendo en cuenta los antecedentes clínicos del paciente, las complicaciones médicas, la edad de presentación y dada la importancia de un diagnóstico preciso para establecer un tratamiento específico, un seguimiento, un pronóstico y un asesoramiento genético, incluido el riesgo de heredabilidad, y dados los hallazgos paraclínicos iniciales que sugieren una cardiopatía congénita de origen genético, se solicitó un estudio molecular de los genes relacionados con la cardiopatía congénita, utilizando la metodología NGS y análisis de CNV (variantes del número de copias).
RESULTADOS
Se realizó la secuenciación del exoma completo (toda la región codificante del genoma) en un secuenciador masivo DNB-SEQ400 o DNBSEQ-T7 de última generación utilizando una biblioteca de exoma MGI-V5. A partir de estos datos, se analizaron los genes relacionados con la cardiopatía congénita. Los genes evaluados alcanzaron una cobertura media superior al 98 % y una profundidad mínima de 20X. No se realizó secuenciación Sanger para confirmación ni para regiones con una cobertura inferior a 20X.
Los genes relacionados con las cardiopatías congénitas que se analizaron fueron: ACTA2, ACTC1, ACVR1, ACVR2B, ACVRL1, ANKRD1, B3GAT3, BMPR2, BRAF, CBL, CFC1, CHD7, CITED2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, CREBBP, CRELD1, DTNA, EFEMP2, EHMT1, ELN, ENG, EP300, EVC, EYA4, FBN1, FBN2, FLNA, FOXC1, FOXF1, FOXH1, FOXP1, GAA, GATA4, GATA5, GATA6, GDF1, GJA1, GJA5, HAND2, HRAS, IRX4, ISL1, JAG1, KANSL1, KCNA5, KCNJ8, KCNK3, KMT2D, KRAS, LEFTY2, MAP2K1, MAP2K2, MCTP2, MED12, MED13L, MFAP5, MIB1, MYBPC3, MYH11, MYH6, MYH7, MYLK, NEXN, NF1, NKX2-5, NKX2-6, NODAL, NOTCH1, NOTCH2, NOTCH3, NPHP4, NRAS, PDGFRA, PITX2, PLOD1, PRKG1, PTPN11, RAF1, RASA1, RASA2, RIT1, SALL4, SHOC2, SKI, SLC2A10, SMAD1, SMAD3, SMAD4, SMAD6, SMAD9, SOS1, SOS2, SPRED1, TAB2, TBX1, TBX20, TBX5, TDGF1, TFAP2B, TGFB2, TGFB3, TGFBR1, TGFBR2, TNNI3, TNNI3K, TOPBP1, UPF3B, ZDHHC9, ZFPM2, ZIC3.
Los resultados de la secuenciación se sometieron a un proceso bioinformático en un análisis secundario para evaluar la calidad de los datos obtenidos de la secuenciación, y en un análisis terciario para alinear las secuencias, realizar la identificación y el filtrado de variantes, siguiendo criterios de calidad específicos. Las variantes se analizaron con respecto al genoma de referencia hg38 para su anotación e identificación.
El análisis se centró en identificar variantes ubicadas en regiones exónicas o de empalme (al menos 20 pb), así como pequeñas inserciones y deleciones. También permitió la detección de deleciones y duplicaciones exónicas, denominadas variantes del número de copias (CNV), y variantes que afectan a grandes regiones génicas, que se notificaron cuando se identificaron. Este método permitió la detección de CNV solo cuando afectaban a más de dos exones.
Todas las variantes identificadas se evaluaron según los criterios de clasificación recomendados por ACMG/AMP (Richards et al., 2015), incorporando las actualizaciones posteriores de ClinGen (clinicalgenome.org). La evaluación incluyó datos de bases de datos establecidas como ClinVar, HGMD, LOVD, dbSNP y gnomAD.
El laboratorio identificó una variante heterocigótica inicialmente clasificada como VUS en el gen MYBPC3 que provoca el cambio de una citosina a una timina en la posición 1433 del ADNc, en el exón 16 del gen (c.1433C>T) y que, a nivel proteico, produce un cambio de sentido de serina a leucina en el aminoácido 478 (p.(Ser478Leu)), un aminoácido moderadamente conservado (Tabla 1).
Tabla 1. Resultados de la secuenciación y análisis de CNV.
| Gen | Variante | Tipo de variante | Ratio alélico VAF | Cigosidad |
| MYBPC3(NM_000256.3) | c.1433C>T p.(Ser478Leu) | Missense | 0.48 | Heterocigoto |
Esta variante aparece registrada en la base de datos ClinVar (ID:180935), clasificada como VUS en 8 entradas, en la base de datos The Human Gene Mutation Database (HGMD) (CM1616589) asociada a miocardiopatía hipertrófica, y en la base de datos Leiden Open Variation Database (LOVD), clasificada como VUS en 3 entradas y como probablemente patógena en 1. En la literatura científica consultada, se ha descrito en dos individuos afectados por miocardiopatía hipertrófica (PMID: 27532257, 29121657) y en tres casos de mortinatalidad (PMID: 30615648). Su frecuencia alélica es de 0,0000288 en la población general (gnomAD). El predictor in silico REVEL la clasifica como variante deletérea (Tabla 2).
Tabla 2. Resumen de casos previamente descritos que portan la misma variante de MYBPC3 y sus características fenotípicas.
| Caso/Referencia (PMID) | Variante (NM_000256.3) | Diagnóstico clínico | Descripción Fenotípica | Edad/Sexo | Resultado y comentarios |
| Viswanathan et al., 2017 (PMID: 29121657) | c.927-9G>A (p.IVS11-9G>A) c.1028delC(p.Thr343MetfsX7) c.1235_1236delTT(p.Phe412Ter) c.2455_2459delATGCG(p.Met819AlafsX12) c.2864_2865delCT(p.Pro955ArgfsX95) c.3192dupC(p.Lys1065GlnfsX12) c.3776delA(p.Gln1259ArgfsX72) | Miocardiopatía hipertrófica | Hipertrofia ventricular izquierda simétrica, función sistólica preservada, penetrancia variable dentro de la familia. No hay correlación clara con el dominio mutado. | 42 ± 17 años (rango 8 meses – 92 años) | De asintomático a HCM severa; fenotipo independiente del sitio de mutación. |
| Dong et al., 2019 (PMID: 30615648) | c.1504C>T (p.Arg502Trp) c.2374C>T (p.Arg792Trp) c.2905G>A (p.Glu969Lys) | Miocardiopatía fetal | Desorganización miocárdica y fibrosis en autopsia, hipertrofia septal, engrosamiento de las paredes ventriculares. | Prenatal / mortinato | Resultado letal; respalda el potencial patogénico en estado homocigoto o compuesto. |
| Field et al., 2022 (PMID: 27532257) | c.1504C>T(p.Arg502Trp) | Miocardiopatía hipertrófica (familiar) | Hipertrofia septal asimétrica, fracción de eyección preservada, herencia autosómica dominante, penetrancia incompleta. | Adultos | Fenotipo leve; se observó segregación familiar. |
| Caso actual (presente informe) | c.1433C>T (p.Ser478Leu) | Cardiopatía congénita con rasgos hipertróficos | Defecto del tabique ventricular con hipertrofia ventricular izquierda concéntrica; no se detectaron otras variantes sarcoméricas. | 5 meses | Sobrevivió; variante clasificada como VUS según ACMG/AMP; se recomienda revaluación. |
Tras un análisis personalizado más detallado que tiene en cuenta todas las herramientas multimodales, la proteína codificada por este gen se conoce como proteína C3 de unión a miosina (de RefSeq NM_000256.3), un gen con ubicación cromosómica 11p11.2. Otra información: transcripción del código genético, ENST00000545968.6.; Código genético: ENSG00000134571.12; posición de transcripción (incluidos los UTR): hg38 chr11: 47 331 406-47 352 702; tamaño: 21 297 kb; posición de la región codificante: hg38 chr11: 47 331 871-47 352 647; tamaño: 20 777 kb; recuento de exones codificantes: 34.
La variante se encuentra dentro de un dominio de inmunoglobulina altamente conservado, en una región previamente asociada con variantes clínicamente relevantes. El análisis de la base de datos (ClinVar, UniProt) reveló un predominio de variantes patógenas en la región circundante, con escasos informes de variantes de significado incierto y ninguna variante benigna. Estos hallazgos respaldan la posible relevancia funcional de la variante identificada y justifican su integración con estudios de correlación fenotípica y segregación familiar para su clasificación adecuada. (Figura 1).
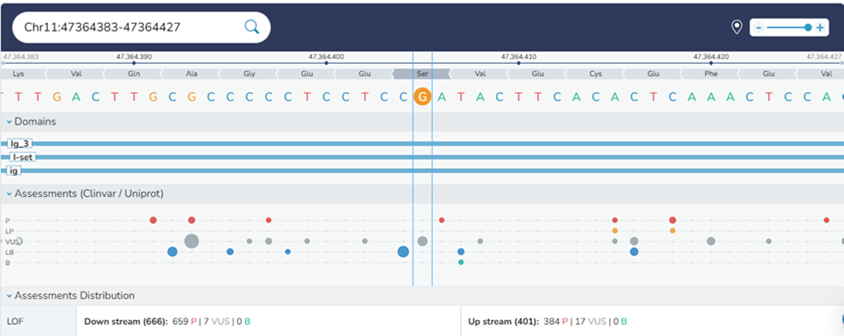
Según UniProtKB/Swiss-Prot, la función molecular del gen MYBPC3 es ser una proteína asociada a los filamentos gruesos situada en la región del puente cruzado de las bandas A del músculo estriado de los vertebrados.
In vitro se une al MHC, a la F-actina y a los filamentos finos nativos, y modifica la actividad de la ATPasa de miosina activada por actina. Puede modular la contracción muscular o desempeñar una función más estructural.
La función molecular-bioquímica del gen MYBPC3, según GENATLAS, consiste en la proteína C3 de unión a la miosina y la titina, expresada en la banda A de los sarcómeros, incluyendo dos isoformas cardíacas y del músculo esquelético rápido, que interactúan con la beta-miosina S2 MYBPC3. Los niveles de expresión génica los tejidos se muestran en la figura 2.
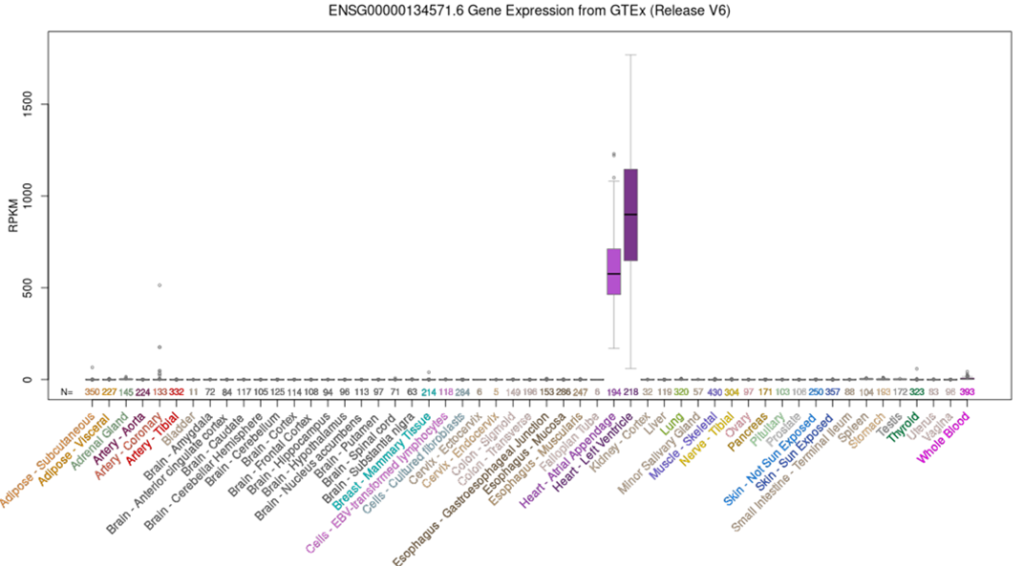
La proteína MYBPC3 (proteína de unión a miosina C, tipo cardíaco) actúa como modulador estructural y funcional del sarcómero cardíaco (figura 3). Su interacción con la miosina y la titina regula tanto la arquitectura como la contractilidad del miocardio. Las variantes patógenas o de significado incierto en este gen pueden alterar la estabilidad del complejo de miosina, alterar la disposición del sarcómero o afectar a la regulación de la contracción, lo que conduce a una disfunción mecánica progresiva. Una variante en MYBPC3 representa un riesgo porque compromete directamente un nodo estructural y funcional esencial en la vía contráctil. Aunque algunas variantes pueden recibir inicialmente una clasificación como variantes de VUS, su ubicación en dominios funcionales conservados, su asociación con fenotipos clínicos (como la miocardiopatía hipertrófica o incluso la cardiopatía congénita) y su potencial efecto disruptivo sobre la dinámica del sarcómero respaldan su relevancia clínica. (Salomonis et al., 2025).
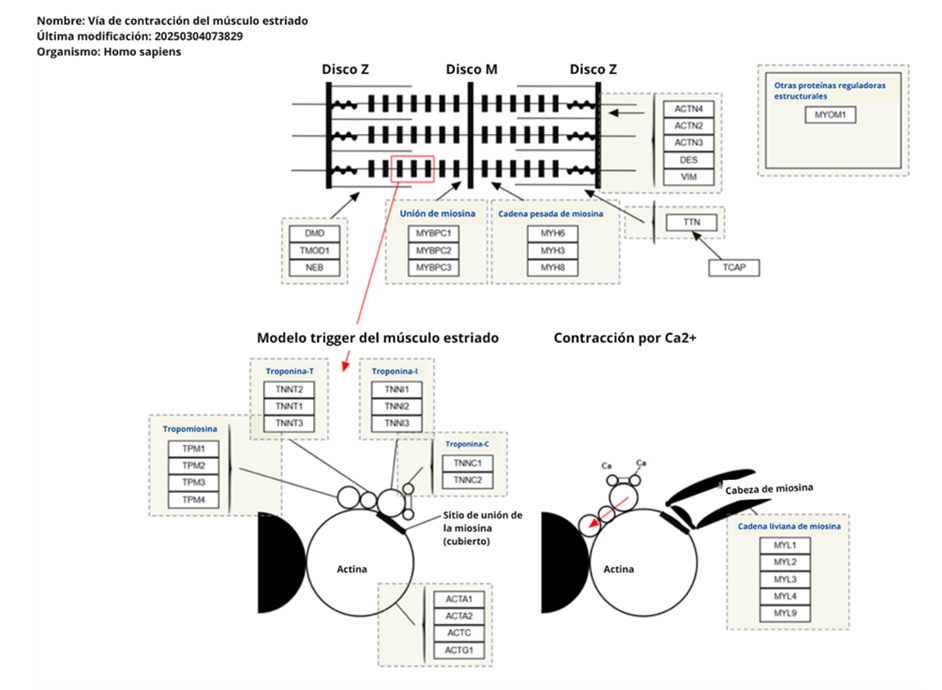
La categorización funcional de MYBPC3 basada en las anotaciones de Gene Ontology muestra su participación en diversos procesos biológicos y funciones moleculares relacionados con la organización del sarcómero, la regulación de la transcripción y el metabolismo celular.
Una búsqueda en la base de datos HPO (Ontología del Fenotipo Humano) documentó que este gen, NCBI Gene:4607, con sinónimos CMD1MM, CMH4, FHC, LVNC10, MYBP-C y cMyBP-C, se ha asociado con 48 fenotipos y tres afecciones médicas. Las características específicas se detallan en las tablas 3 y 4.
Tabla 3. Afecciones relacionadas con el gen MYBPC3.
| Enfermedad | OMIM | ORPHA | MONDO | Herencia | Rasgos Clínicos |
| No compactación del ventrículo izquierdo 10 | 615396 | 54260 | MONDO:0014163 | Autosómica dominante | Insuficiencia cardiaca congestiva, hipertensión arterial pulmonar, síncope, aumento del volumen telediastólico del ventrículo izquierdo, miocardiopatía dilatada |
| Miocardiopatía hipertrófica familiar, tipo 4 | 115197 | 99739 | MONDO:0007268 | Autosómica dominante | Miocardiopatía hipertrófica neonatal grave |
| Miocardiopatía dilatada familiar aislada | 15200 | 154 | MONDO:0700335 | Autosómica dominante, autosómica recesiva, herencia mitocondrial, recesiva ligada al X | Fracción de eyección del VI menor del 25% y/o fracción de eyección del VI menor del 45% con diámetro telediastólico del VI mayor del 117% del valor predicho |
Tabla 4. Rasgos fenotípicos relacionados con el gen MYBPC3.
| HPO | Rasgo | HPO | Rasgo |
| HP:0025169 | Disfunción sistólica del ventrículo izquierdo | HP:0011462 | Inicio adulto joven |
| HP:0001297 | Accidente cerebrovascular | HP:0012764 | Ortopnea |
| HP:0001279 | Sïncope | HP:0003198 | Miopatía |
| HP:0000007 | Herencia autosómica recesiva | HP:0030718 | Agrandamiento de la aurícula derecha |
| HP:0000006 | Herencia autosómica dominante | HP:0011623 | Defecto septal ventricular muscular |
| HP:0033755 | Aumento del volumen telediastólico del ventrículo izquierdo | HP:0000969 | Edema |
| HP:0002098 | Distrés respiratorio | HP:0011675 | Arritmia |
| HP:0002094 | Disnea | HP:0005144 | Hipertrofia septal ventricular izquierda |
| HP:0002092 | Hipertensión pulmonar | HP:0031318 | Desorden de miofibras cardíacas |
| HP:0100578 | Lipoatrofia | HP:0002875 | Disnea de esfuerzo |
| HP:0011712 | Bloqueo de rama derecha | HP:0001541 | Ascitis |
| HP:0011713 | Bloqueo de rama izquierda | HP:0012378 | Fatiga |
| HP:0100598 | Edema pulmonar | HP:0001695 | Paro cardíaco |
| HP:0011705 | Bloqueo auriculoventricular de primer grado | HP:0001698 | Derrame pericárdico |
| HP:0003457 | Anomalía del EMG | HP:0001678 | Bloqueo auriculoventricular |
| HP:0002240 | Hepatomegalia | HP:0001645 | Muerte súbita cardíaca |
| HP:0003584 | Inicio tardío | HP:0001644 | Miocardiopatía dilatada |
| HP:0003581 | Inicio adulto | HP:0001663 | Fibrilación ventricular |
| HP:0100749 | Dolor de pecho | HP:0001640 | Cardiomegalia |
| HP:0002326 | Ataque isquémico transitorio | HP:0001639 | Miocardiopatía hipertrófica |
| HP:0003623 | Inicio neonatal | HP:0001635 | Insuficiencia cardíaca congestiva |
| HP:0003621 | Inicio juvenil | HP:0001727 | Accidente cerebrovascular tromboembólico |
| HP:0012664 | Fracción de eyección reducida | HP:0000407 | Discapacidad auditiva/hipoacusia neurosensorial |
| HP:0030682 | Ventriculo izquierdo no compactado | HP:0001714 | Hipertrofia ventricular |
La variante se encuentra en un gen para el que existen directrices específicas de ClinGen (Especificaciones del panel de expertos de ClinGen para las directrices de interpretación de variantes ACMG/AMP para MYBPC3, versión 1.0.0).
De acuerdo con las directrices ACMG/AMP y las especificaciones de Clingen, esta variante se clasifica como de patogenicidad tipo PM2, PP3, con software que respalda y demuestra el efecto sobre la proteína, datos computacionales (VARITY, Revel, predicción agregada), frecuencias poblacionales (<0,01 %: gnomAD (máx.), gnomAD (agregado), gnomAD (genoma), ExAC, gnomAD (exoma) y < 0,02 % 4,7KJPN), datos biológicos, datos funcionales in vitro, datos de predicción estructural en un paciente con presentación en la infancia con un historial probablemente relacionado en el que, según la sensibilidad a la dosis de ClinGen, hay pruebas suficientes que respaldan la haploinsuficiencia como mecanismo patogénico. Esto implica que las variantes con pérdida de función, como las que producen una proteína truncada y conducen a la expresión de solo el alelo restante, son causantes de la enfermedad. Si bien la variante aquí descrita no da lugar a una proteína truncada, su impacto potencial debe interpretarse a la luz de esta sensibilidad a la dosis establecida.
DISCUSIÓN
La comprensión actual de la miocardiopatía hipertrófica (MCH) ha evolucionado considerablemente gracias a los análisis genéticos de alta resolución, que han identificado el gen MYBPC3 como el principal factor contribuyente en una proporción significativa de casos. La mayoría de las variantes patógenas son truncadas, lo que conduce a una haploinsuficiencia proteica y a una alteración directa del ensamblaje sarcomérico, produciendo un estado de hipercontractilidad y relajación deficiente, el sello distintivo de la fisiopatología de la MCH (Toepfer et al., 2019).
Aunque las variantes de MYBPC3 se relacionaron inicialmente con fenotipos leves y de aparición tardía, estudios pediátricos recientes han revelado manifestaciones tempranas y heterogéneas (Ananthamohan et al., 2024). Field y colaboradores (2022) informaron de que más del 14% de los pacientes menores de 18 años con variantes patógenas de MYBPC3 experimentaron eventos adversos graves, como muerte súbita y arritmias ventriculares graves. De manera similar, en nuestro paciente, el inicio clínico se produjo antes del año de edad y se asoció a un fenotipo estructural complejo, lo que respalda el concepto de que la carga alélica y la interacción de las variantes pueden modular la gravedad de la enfermedad (Patiño Moncayo y Nieto Zárate, 2024)
Ashraf y colaboradores (2025) demostraron además una amplia variabilidad intrafamiliar en los fenotipos relacionados con MYBPC3, desde hipertrofia leve hasta miocardiopatía obstructiva con insuficiencia cardíaca. Esta variabilidad refleja el presente caso, en el que no se observó miocardiopatía manifiesta, pero se documentó un defecto cardíaco congénito complejo (ASD, VSD, PDA, reconstrucción de la arteria pulmonar), lo que amplía el espectro fenotípico de MYBPC3 hacia malformaciones del desarrollo.
La identificación de la variante c.1433C>T (p.Ser478Leu) refuerza las pruebas emergentes que implican a los genes sarcoméricos en la morfogénesis cardíaca temprana.
Tudurachi y colaboradores (2023) demostraron que casi el 50 % de las variantes patógenas del gen MYBPC3 dan lugar a proteínas truncadas que se degradan por la degradación mediada por nonsense (NMD) o las vías de ubiquitina-proteasoma, mientras que Chiswell y colaboradores (2023) destacaron las disfunciones contráctiles tempranas que afectan a la formación de las cavidades embrionarias.
Sierant y colaboradores (2025) han demostrado que las variantes heterocigotas en los genes sarcoméricos representan hasta el 10 % de los casos de cardiopatías congénitas aisladas, que van desde defectos leves hasta complejos, lo que concuerda con el fenotipo de nuestro paciente. De manera similar, Dong y colaboradores (2025) describieron variantes bialélicas del gen MYBPC3 que contribuyen a la cardiopatía congénita incluso en familias no consanguíneas, lo que destaca la importancia de explorar las variantes heterocigotas en presentaciones congénitas no letales.
En consonancia con las directrices actuales (Abbas et al., 2024), este caso subraya el valor diagnóstico de los paneles de secuenciación de nueva generación (NGS) tanto en las miocardiopatías como en las cardiopatías congénitas, con un rendimiento de hasta el 60 % en los casos familiares. Además, las variantes de VUS, como la identificada aquí, no deben descartarse, sino integrarse en el marco etiológico cuando existan correlaciones clínicas, bioinformáticas y familiares consistentes (Richards et al., 2015; Lee et al., 2025).
En nuestro paciente, el fenotipado reverso y el análisis multimodal revelaron una concordancia entre la función biológica del gen y los hallazgos clínicos observados, lo que ilustra el valor práctico de la interpretación dinámica de variantes. Desde el punto de vista funcional, tanto las variantes truncadas como las no truncadas pueden producir resultados clínicos similares, lo que respalda un mecanismo de pérdida de función independiente del locus (Helms et al., 2020). Además, la deficiencia de MYBPC3 activa las vías fibróticas y de remodelación a través de la señalización NF-κB, TGF-β1 y HIF-1α (Zou et al., 2022), e influye en la regulación transcripcional y metabólica de los cardiomiocitos (Chen et al., 2025).
Desde el punto de vista clínico, nuestros hallazgos coinciden con los de Park y colaboradores (2022), quienes destacaron el infradiagnóstico de los portadores de variantes patógenas de MYBPC3 sin CMH manifiesta, pero con hallazgos ecocardiográficos sugestivos. Esto refuerza la necesidad de realizar un cribado genético sistemático en las cardiopatías estructurales pediátricas.
Según las especificaciones del Panel de Expertos en Cardiomiopatía de ClinGen (CMCEP), la variante c.1433C>T (p.Ser478Leu) cumple los criterios PM2 (frecuencia poblacional muy baja) y PP3 (evidencia computacional deletérea) de ACMG/AMP. Además, MYBPC3 tiene una sensibilidad de haploinsuficiencia de nivel 3 en el mapa de sensibilidad a la dosis de ClinGen, lo que respalda su posible papel causal, incluso aunque esté clasificado como VUS.
Aunque no se pudo realizar un análisis de segregación familiar, esta sigue siendo un elemento clave para discernir la patogenicidad de la variante. La ausencia de dicho análisis impide la confirmación definitiva de una relación causal. Sin embargo, el fenotipo clínico consistente, la plausibilidad biológica y las predicciones de patogenicidad in silico sugieren conjuntamente un probable papel contribuyente de esta variante en el fenotipo cardíaco del paciente.
En general, este caso respalda un continuo genético y fenotípico que vincula la disfunción sarcomérica con la cardiopatía estructural congénita, lo que destaca la importancia de la evaluación genómica integral, el seguimiento longitudinal y la interpretación iterativa de las variantes en la cardiogenética pediátrica.
CONCLUSIONES
La integración de herramientas genómicas multimodales en el estudio de las cardiopatías congénitas permite comprender mejor la etiología de estos defectos, en particular al revelar la implicación de genes sarcoméricos tradicionalmente relacionados con las miocardiopatías adquiridas. El análisis de variantes en genes como MYBPC3, bajo marcos de interpretación específicos y estandarizados, como las directrices ACMG/AMP adaptadas por ClinGen, demuestra que la contribución genética a las malformaciones cardíacas estructurales puede subestimarse cuando se aplican criterios convencionales sin contextualización funcional o fenotípica.
La presencia de variantes de significado incierto en genes con expresión cardíaca temprana y función estructural crítica subraya la necesidad de tener en cuenta el contexto clínico, biológico y evolutivo en su interpretación. La combinación de predictores in silico, datos de expresión tisular, evidencia funcional y correlación fenotípica representa un enfoque sólido para evaluar su posible contribución patogénica, incluso en ausencia de una clasificación definitiva.
La implementación de estrategias de diagnóstico que incorporan paneles ampliados, fenotipado inverso y criterios de interpretación genética dinámica refuerza la medicina personalizada y de precisión. Este enfoque no solo mejora la detección de las causas subyacentes, sino que también optimiza el seguimiento, la estratificación del riesgo y el asesoramiento familiar, lo que tiene un impacto directo en la práctica clínica y el diseño de futuras investigaciones en la búsqueda de la medicina de precisión.
Declaración de conflicto de intereses
Los autores declaran que no tienen ningún conflicto de intereses en relación con la publicación de este manuscrito.
No se ha recibido financiación para la redacción o publicación de este manuscrito.
Aspectos éticos
El estudio fue aprobado por el comité de ética institucional. Se obtuvo el consentimiento informado por escrito para participar del tutor legal del paciente.
Se obtuvo el consentimiento informado por escrito para la publicación del caso y los hallazgos genéticos acompañantes del tutor legal del paciente.
Disponibilidad de datos y material
Todos los datos que respaldan los hallazgos de este estudio están disponibles a través del autor correspondiente previa solicitud razonable.
Contribución de los autores
Mariana Pérez: Conceptualización, redacción del borrador original, revisión y edición.
Lina Moreno: Visualización, recursos, supervisión, revisión y edición.


