
INTRODUCCIÓN
Se estima que existen más de 1100 enfermedades hereditarias recesivas, las cuales son infrecuentes de forma individual pero suponen un gran número de afectos en su conjunto (Bell et al., 2011). Asimismo, se considera que aproximadamente 1-2 parejas de cada 100 están en riesgo de tener un hijo afecto por una de estas enfermedades (Ropers, 2012). Cuando ambos miembros de una pareja son portadores de la misma enfermedad recesiva, tienen un riesgo de un 25% de tener un hijo afecto en cada gestación. Los individuos portadores no suelen tener antecedentes personales o familiares relacionados con la enfermedad, por lo que desconocen la existencia de estos riesgos. Gracias a los rápidos y constantes progresos en el campo de la genética reproductiva, disponemos de las técnicas necesarias para poder identificar a estas parejas y, de esta forma, reducir los riesgos de que puedan transmitir estas enfermedades.
En el ámbito de la medicina reproductiva resulta fundamental poder identificar a portadores de este tipo de enfermedades, no sólo en parejas con deseos reproductivos, sino también en candidatos a ser donantes de gametos. La donación de gametos es una de las técnicas de reproducción asistida más empleadas en la actualidad. Según los datos recogidos por la Sociedad Española de Fertilidad (SEF), durante el año 2015 se realizaron en España más de 14.000 ciclos de recepción de ovocitos, lo que supone el 11,5% de los ciclos totales de fecundación in vitro, y más de 20.000 ciclos utilizando semen de donante (SEF, 2015).
La legislación vigente referente a donación de gametos, tanto a nivel nacional (Ley 14/2006 y Real Decreto-ley 9/2014) como europeo (Directiva 2004/23/CE), establece las normas de calidad y seguridad que deben seguir los centros autorizados durante el proceso de donación. Entre estas normas, se contemplan los criterios de selección y evaluación clínica de los donantes de células reproductoras. Respecto a los estudios genéticos, se indica que se debe llevar a cabo un cribado genético de genes recesivos prevalentes en la etnia del donante en función de los datos científicos internacionales, así como una valoración del riesgo de transmisión de enfermedades hereditarias conocidas y presentes en la familia. No obstante, en ninguna de estas normativas se definen qué pruebas genéticas concretas deben realizarse, por lo que se deja a criterio de los centros de reproducción la selección de las mismas.
Dada la controversia generada a partir de esta legislación, la SEF y la Asociación para el Estudio de la Biología de la Reproducción (ASEBIR) publicaron en el año 2012 un documento conjunto de recomendaciones para la aplicación del RD1310/2006 (Alonso et al., 2012). En este documento se recomienda evaluar los antecedentes personales y familiares de enfermedades hereditarias, por lo que se identificarían las enfermedades de herencia dominante, estudiar el cariotipo a todos los donantes y analizar las enfermedades recesivas de mayor prevalencia según la etnia. Además, se deja abierta la posibilidad de incluir otras enfermedades genéticas de acuerdo al estado del conocimiento y al desarrollo tecnológico.
En los últimos años, la aparición de las nuevas tecnologías de secuenciación masiva conocidas como Next-Generation Sequencing (NGS) ha supuesto una revolución en el campo de la genética clínica, permitiendo desarrollar nuevas estrategias diagnósticas y ofreciendo la posibilidad de estudiar un gran número de genes de forma simultánea y en un tiempo y coste razonables (Metzker, 2010). Los avances en NGS han promovido el desarrollo de múltiples test genéticos diseñados con diferentes objetivos. Su uso se ha extendido recientemente a ámbitos como la genética reproductiva, donde se emplea de forma preconcepcional en los programas de cribado de portadores tanto para parejas con deseos reproductivos como para donantes de gametos, con la finalidad de poder disminuir el riesgo de transmitir enfermedades hereditarias a la descendencia (Martin et al., 2015; Abulí et al., 2016).
Dadas las ventajas que ofrece la tecnología NGS, en los test de cribado de portadores diseñados en los últimos años se incluyen cientos de enfermedades, por lo que son conocidos como test de cribado ampliado de portadores. Actualmente, existe en el mercado una extensa variedad de este tipo de test que se diferencian en el conjunto de enfermedades que estudian. Estas diferencias radican tanto en el número como en el tipo de enfermedades, la edad a la que se inician, posibilidad de tratamiento y severidad (Henneman et al., 2016). Varias sociedades científicas internacionales han reaccionado ante esta falta de consenso elaborando guías con criterios para la selección de estas enfermedades (Edwards et al., 2015; Henneman et al., 2016; ACOG, 2017). Las recomendaciones apuntan a que los trastornos incluidos en los test de cribado ampliado de portadores deben ser graves, de inicio temprano, que tengan asociadas variantes patogénicas con alta penetrancia y con una clara relación genotipo-fenotipo.
El objetivo de este trabajo es presentar los resultados obtenidos a partir del desarrollo de un test de cribado genético de portadores, diseñado siguiendo las últimas recomendaciones de las sociedades científicas nacionales e internacionales, para que sirva de propuesta para armonizar los estudios genéticos en donantes en los centros de reproducción y en bancos de gametos. Este test se ha empleado como método de cribado de portadores de las enfermedades autosómicas recesivas y ligadas a X que, cumpliendo con los criterios de dichas sociedades, son más prevalentes en nuestro medio, en candidatos a donantes de gametos mediante tecnología NGS. Se presentan los datos de los primeros resultados.
MATERIALES Y MÉTODOS
Población de estudio
Este estudio retrospectivo ha sido llevado a cabo a partir de los resultados del cribado genético de donantes de gametos realizado desde Abril de 2016 hasta Octubre de 2017 en la Unidad de Genética del Hospital HLA Vistahermosa de Alicante con la colaboración de E-GENETICARE Consejo Genético.
Se han incluido 523 individuos candidatos a ser donantes de gametos procedentes de diversos centros españoles de reproducción asistida. Los candidatos a donantes fueron seleccionados en los correspondientes centros siguiendo la legislación vigente (Ley 14/2006 y Real Decreto-ley 9/2014). De este modo, los donantes tenían edades comprendidas entre 18 y 35 años, presentaban buena salud psicofísica y no tenían antecedentes familiares o personales de enfermedades genéticas hereditarias ni alteraciones en el cariotipo. En todos los casos se firmó y recogió un consentimiento informado específico por parte de los centros de reproducción, que también fueron los encargados de realizar el asesoramiento genético a los candidatos. El consentimiento informado fue elaborado siguiendo las recomendaciones de la Asociación Española de Genética Humana (AEGH) (Pampols et al., 2013).
A los candidatos que fueron rechazados del programa de donación debido al resultado del estudio genético se les proporcionó asesoramiento genético post-test cuando así lo habían requerido en la sesión pre-test.
Del total de individuos, 402 (76.9%) eran mujeres y 121 (23.1%) varones. La población objeto de estudio era de etnia caucásica.
Extracción del ADN genómico
El ADN genómico fue extraído a partir de leucocitos de sangre periférica utilizando el kit Cell SV Mini (GeneAll) siguiendo las indicaciones del mismo. La cantidad y calidad del ADN extraído fue determinada mediante el equipo Qubit 2.0 Fluorometer (Thermo Scientific).
Diseño del panel de genes
La selección de genes incluidos en el panel de cribado de donantes de gametos se realizó siguiendo las recomendaciones establecidas por las sociedades científicas European Society of Human Genetics (ESHG) (Henneman et al., 2016), American College of Medical Genetics and Genomics (ACMG), American College of Obstetricians and Gynecologists (ACOG), National Society of Genetic Counselors (NSGC), Perinatal Quality Foundation y Society for Maternal-Fetal Medicine (SMFM) (Edwards et al., 2015).
En base a estos criterios, se seleccionaron 15 genes asociados a 16 enfermedades autosómicas recesivas y ligadas a X incluidas en la base de datos OMIM (Online Mendelian Inheritance Man, www.ncbi.nlm.nih.gov/omim) (Tabla 1). Estas enfermedades tienen como características comunes que presentan una elevada prevalencia (Lazarin et al., 2013), son enfermedades graves de aparición temprana y tienen un clara relación genotipo-fenotipo. Uno de los genes incluidos en el panel, el gen HBB, está relacionado tanto con la anemia falciforme como con la beta-talasemia.
Dado que el síndrome de X-frágil presenta un tipo de herencia dominante ligado a X, el estudio de la expansión del triplete CGG de la región promotora del gen FMR1 se realizó únicamente en las mujeres de la población de estudio.
Secuenciación masiva
El panel de genes se desarrolló mediante tecnología de amplicones empleando la plataforma de NGS GeneRead v2 (Qiagen) y posterior secuenciación mediante un equipo Miniseq (Illumina).
Análisis de resultados
Se consideraron las variantes con un número de lecturas superior a 50 y con una frecuencia superior al 20% en los SNP y del 25% en los INDELs. Se tuvieron en cuenta las regiones codificantes más 10 pb de los intrones flanqueantes. Se consideraron polimorfismos aquellos cambios que presentan una frecuencia en la población superior al 5%.
El alineamiento de la secuencia se realizó empleando la herramienta BWA. Para el variant calling se ha utilizado la herramienta GATK Unified Genotyper.
Atendiendo a las recomendaciones de ACMG, ACOG, NSGC, Perinatal Quality Foundation y SMFM, no se reportaron las variantes de significado desconocido (VUS) (Edwards et al., 2015). Únicamente se consideraron las variantes clasificadas como patogénicas o probablemente patogénicas según la base de datos ClinVar.
Por último, siempre se indicó el riesgo residual para cada gen estudiado.
Técnicas complementarias
El análisis del número de copias del exón 7 del gen SMN1 se llevó a cabo mediante qPCR y MLPA (salsa P060, MRC-Holland, Amsterdam, Holanda). La expansión del triplete CGG de la región promotora del gen FMR1 se analizó mediante TP-PCR (FRAXA1, Experteam) seguida de análisis de fragmentos.
RESULTADOS
Implementación del cribado genético mediante un panel de 15 genes en donantes de gametos
En el estudio se incluyeron 523 individuos candidatos a ser donantes de gametos: 402 (76.9%) mujeres y 121 (23.1%) varones. El cribado genético se llevó a cabo mediante el análisis de un panel de 15 genes asociados a 16 enfermedades autosómicas recesivas y ligadas a X mediante tecnología NGS (Tabla 1). Las enfermedades seleccionadas son graves, de aparición temprana, con una clara relación genotipo-fenotipo y presentan una elevada prevalencia en nuestro medio (Lazarin et al., 2013).
La población objeto de estudio procedió de distintos centros españoles de reproducción asistida, los cuales solicitaron realizar un cribado genético a los candidatos con el objetivo de conocer su estatus de portadores y, de esta forma, poder asignarlos a un individuo que no es portador de ninguna mutación en el mismo gen en el caso de las enfermedades recesivas (matching genético) o descartarlos del programa de donación de gametos en el caso de las donantes portadoras de enfermedades ligadas al cromosoma X.
Resultado del cribado genético
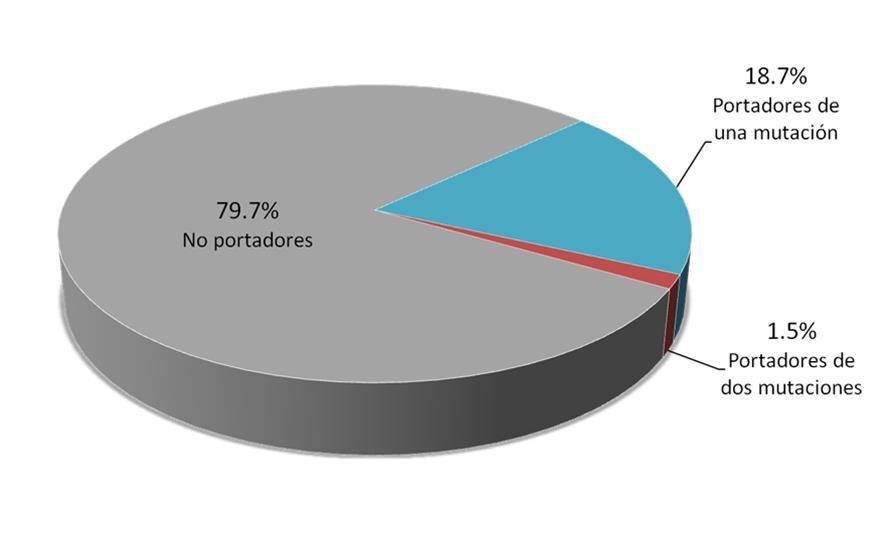
A partir del cribado genético realizado en la población objeto de estudio, se observó que un 79.7% (417/523) de individuos no presentaron ninguna mutación en los genes incluidos en el panel, mientras que un 20.3% (106/523) de ellos eran portadores de al menos una mutación patogénica o probablemente patogénica (Figura 1). Del total de individuos estudiados, un 18.7% (98 individuos) presentó una única mutación patogénica, mientras que en un 1.5% (8 individuos) se identificaron dos mutaciones patogénicas en genes distintos.
La tabla 1 muestra la frecuencia de portadores obtenida a partir de este estudio para cada enfermedad, siendo las que presentan mayores tasas de portadores: fibrosis quística (4.6%), sordera congénita no sindrómica (3.6%), atrofia muscular espinal (2.1%), fenilcetonuria (1.7%) y fiebre mediterránea familiar (1.5%). Entre todas las mutaciones patogénicas o probablemente patogénicas identificadas en la población estudiada, la más frecuente fue la mutación p.Phe508del del gen CFTR (12 individuos). Se identificaron 17 portadores del alelo 5T, por lo que teniendo en cuenta dicho alelo, la tasa de portadores de mutación en el gen CFTR sería de 7.8% (Tabla 1).
Cabe mencionar que entre las mujeres de la población de estudio, se identificaron 4 portadoras de un alelo del gen FMR1 en la zona intermedia o zona gris, es decir, un alelo con 45 a 54 repeticiones del triplete CGG en la región promotora del gen (Tabla 1).
| Tabla 1: Frecuencia de portadores para las enfermedades estudiadas en el presente estudio. Se indica el gen asociado a cada una de ellas y el patrón de herencia. | ||||
| Enfermedad (número OMIM) | Gen | Tipo de herencia | Frecuencia de portadores | |
| N (%) | 1 en _ | |||
|
Fibrosis quística (#602421) |
CFTR | AR | 24 (4.6%)a | 22 |
|
Sordera congénita no sindrómica (DFNB1) (#220290) |
GJB2 | AR | 19 (3.6%) | 28 |
|
Atrofia Muscular Espinal (#253300) |
SMN1 | AR | 11 (2.1%) | 48 |
|
Fenilcetonuria (#261600) |
PAH | AR | 9 (1.7%) | 58 |
|
Fiebre Mediterránea Familiar (#249100) |
MEFV | AR | 8 (1.5%) | 65 |
|
Hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa (#201910) |
CYP21A2 | AR | 6 (1.1%) | 87 |
|
Síndrome de Smith-Lemli-Opitz (#270400) |
DHCR7 | AR | 5 (1.0%) | 105 |
|
Deficiencia de acil- CoA deshidrogenada (cadena media) (#607008) |
ACADM | AR | 5 (1.0%) | 105 |
|
Síndrome de X-Frágil (#300624) |
FMR1 | LX | 4 (1.0%)b | 101 |
|
Anemia falciforme (#603903) |
HBB | AR | 2 (0.4%) | 262 |
|
Enfermedad de Canavan (#271900) |
ASPA | AR | 1 (0.2%) | 523 |
|
Enfermedad de Gaucher (#230800) |
GBA | AR | 1 (0.2%) | 523 |
|
Enfermedad de Pompe (#232300) |
GAA | AR | 1 (0.2%) | 523 |
|
Beta-talasemia (#613985) |
HBB | AR | 1 (0.2%) | 523 |
|
Enfermedad de Tay-Sachs (#272800) |
HEXA | AR | 0 | – |
|
Síndrome de Riley-Day (Disautonomía Familiar) (#223900) |
IKBKAP | AR | 0 | – |
| a Número de portadores de mutación patogénica en el gen CFTR, sin considerar el alelo 5T. Si se tiene en cuenta que se identificaron 17 individuos portadores de este alelo, la frecuencia de portadores de fibrosis quística sería de 7.8%.
b Número de portadoras de un alelo en la zona intermedia o zona gris (45-54 repeticiones del triplete CGG de la región promotora del gen FMR1). Este estudio se realizó únicamente en población femenina (n=402). (AR) Herencia autosómica recesiva. (XL) Herencia ligada a X. (N) Número de portadores. |
||||
DISCUSIÓN
El presente estudio muestra los resultados de la implementación de un test de cribado de portadores en una población de candidatos a donantes de gametos. El test consiste en un panel de 15 genes asociados a 16 enfermedades autosómicas recesivas y ligadas a X analizado mediante NGS.
El propósito principal de los test de cribado de portadores es identificar a aquellos individuos o parejas que se encuentran en riesgo de transmitir una enfermedad hereditaria y, de esta forma, tener la posibilidad de conocer las opciones reproductivas disponibles, tomar decisiones informadas y reducir este riesgo (Henneman et al., 2016; Rose et al., 2016). La tecnología NGS permite estudiar un gran número de genes de forma coste-efectiva, lo que ha promovido que en los test de cribado ampliado de portadores se incluyan cientos de enfermedades hereditarias. No obstante, cabe tener en cuenta que a medida que aumenta el número de genes estudiados también se incrementa la complejidad y el tiempo del estudio, así como la dificultad del manejo clínico de la información obtenida, debido principalmente a la detección de un mayor número de variantes de significado incierto (VUS). También se debe valorar en qué medida los resultados obtenidos a partir de los cribados ampliados de portadores alteran el manejo clínico de las parejas con deseos reproductivos. En este sentido, el estudio realizado por Franasiak et al. en parejas infértiles mostró que la implementación de este tipo de cribado afecta a las decisiones clínicas únicamente en un 0.21% de los casos (Franasiak et al., 2016). Además, determinados test tienen una utilidad limitada en la práctica clínica, pues incluyen enfermedades que no se consideran graves, tienen baja penetrancia o se inician en la vida adulta y, por lo tanto, las sociedades científicas no recomiendan incluirlas (Edwards et al., 2015; Henneman et al., 2016; ACOG, 2017). Algunos ejemplos de estas enfermedades son la deficiencia de alfa-1 antitripsina y la hemocromatosis hereditaria (Wienke et al., 2014; Edwards et al., 2015). Estos aspectos pueden llegar a generar incertidumbre y ansiedad tanto a los donantes como a los pacientes respecto a sus riesgos reproductivos, lo que debe evitarse en la práctica clínica (Dondorp et al., 2014). Por lo tanto, a la hora de diseñar los test de cribado de portadores es fundamental buscar un equilibrio entre el número de enfermedades incluidas y su utilidad clínica.
Este estudio se ha llevado a cabo en una población de 523 candidatos a donantes de gametos, de los cuales un 20.3% ha resultado ser portador de al menos una mutación patogénica o probablemente patogénica en los genes estudiados. Esta tasa de portadores varía entre los estudios reportados en la bibliografía debido a las diferencias en cuanto al tamaño muestral, el número de enfermedades estudiadas y la tecnología empleada en el análisis. Lazarin et al. realizaron un estudio de cribado de portadores en 23,453 individuos, mediante análisis de genotipado de 417 mutaciones asociadas a 108 enfermedades recesivas, y observaron que el 24.0% de los individuos eran portadores de al menos una mutación (Lazarin et al., 2013). Más recientemente, Martin et al. llevaron a cabo un cribado ampliado de portadores empleando un panel de 549 genes analizados mediante NGS en 2,570 individuos, de los cuales el 84.1% resultaron ser portadores de al menos una variante patogénica (Martin et al., 2015). Abulí et al. también emplearon esta tecnología para realizar un cribado de portadores en una población de 1,301 individuos, mediante un panel de 200 genes asociados a 368 enfermedades, y detectaron que el 56.3% de los individuos eran portadores de al menos una mutación patogénica (Abulí et al., 2016). Como se puede observar a partir de estos datos, las tasas de portadores detectadas no aumentan de forma proporcional al número de genes estudiados, lo que probablemente es debido a que en los test de portadores se incluyen enfermedades cuyas frecuencias de portadores son extremadamente bajas.
Respecto a las tasas de portadores de cada enfermedad, dadas las diferencias en el número de individuos estudiados nuestros resultados no son comparables con los reportados en la bibliografía. No obstante, cabe mencionar que las enfermedades con mayores tasas de portadores detectadas a partir de este estudio (fibrosis quística, sordera congénita no sindrómica, atrofia muscular espinal, fenilcetonuria y fiebre mediterránea familiar) también se encuentran entre las enfermedades con mayores frecuencias de portadores en los estudios indicados anteriormente (Lazarin et al., 2013; Abulí et al., 2016). En particular, la fibrosis quística fue la enfermedad para la que se obtuvo una mayor tasa de portadores (4.6%). Si se tiene en cuenta el número de portadores del alelo 5T (17 individuos), la frecuencia de portadores de fibrosis quística ascendería a 7.8%. El alelo 5T del gen CFTR se ha asociado a formas no clásicas de la enfermedad y a agenesia bilateral de los conductos deferentes (Groman et al., 2004), por lo que consideramos que debe formar parte del cribado de portadores en los programas de donación de gametos.
En la población de este estudio únicamente se han incluido candidatos a donantes de gametos cuyo historial clínico personal y familiar había sido previamente evaluado, como exige la legislación española, lo que permitió identificar y descartar a candidatos procedentes de familias con enfermedades dominantes. Los candidatos que fueron identificados como portadores de una enfermedad recesiva a partir de este cribado no fueron descartados del proceso de donación, sino que se realizó un matching genético, es decir, fueron emparejados con un individuo que no era portador de ninguna mutación en el mismo gen, tal y como recomienda ASEBIR (ASEBIR, 2016). Sin embargo, en el caso de las portadoras de una mutación asociada al síndrome de X-frágil, sí fueron descartadas del programa de donación de ovocitos, puesto que la enfermedad tiene un patrón de herencia dominante ligada al cromosoma X. Se identificaron 4 portadoras de alelos intermedios de 45-54 repeticiones del triplete CGG de la región promotora del gen FMR1 entre la población estudiada (402 mujeres). Los alelos intermedios no están clasificados como causantes del síndrome de X-frágil. No obstante, se ha reportado que alrededor del 14% son inestables y cuando se transmiten vía materna pueden expandirse hasta la zona de premutación (Nolin et al., 2011), que está asociada a riesgos aumentados de fallo ovárico precoz y síndrome de temblor/ataxia asociado a X-frágil (FXTAS). Por ello, nuestra recomendación es excluir a estas mujeres del programa de donación de ovocitos.
Una de las limitaciones de este test de cribado es que se han utilizado riesgos pan-étnicos, como es la tendencia actual debido a la movilidad de la población (Lazarin et al., 2013). En el caso de algunos grupos étnicos, como los judíos askenazí, sería recomendable utilizar un panel más específico.
Como conclusión, el presente estudio muestra los resultados del diseño e implementación de un test de cribado de portadores analizado mediante NGS, con un número pequeño de genes a estudiar y aplicado a candidatos a donación de gametos. El test se ha diseñado siguiendo las recomendaciones de las sociedades científicas, por lo que se han seleccionado 15 genes asociados a 16 enfermedades autosómicas recesivas y ligadas a X caracterizadas por ser graves, de aparición temprana, con una clara relación genotipo-fenotipo y con una elevada prevalencia. El test diseñado facilita el manejo clínico de la prueba y potencia su utilidad clínica, permitiendo reducir los riesgos de transmitir las enfermedades hereditarias recesivas más prevalentes en nuestro medio y favoreciendo la autonomía en las decisiones reproductivas de las parejas para tener un hijo sano.
AGRADECIMIENTOS
Este trabajo ha sido subvencionado parcialmente por E-GENETICARE S.L. y Citolab S.L.
