Pilar Rivero-Ríos1 y Sabine Hilfiker1
1Instituto de Parasitología y Biomedicina López-Neyra (CSIC), Granada, España
La enfermedad de Parkinson (EP) es una enfermedad neurodegenerativa caracterizada por la pérdida de neuronas dopaminérgicas en la substancia nigra y por la presencia de agregados proteicos, conocidos como cuerpos de Lewy, en las neuronas que sobreviven. La existencia de estos agregados proteicos es indicativa de que defectos en la ruta autofágica podrían contribuir a la enfermedad.
La autofagia es el proceso mediante el cual proteínas, agregados proteicos y orgánulos disfuncionales son transportados al lisosoma para su degradación. La macroautofagia es la forma más común de autofagia y comienza con la formación de una estructura de doble membrana denominada fagóforo, que se va elongando y secuestrando en su interior componentes citoplasmáticos, para concluir con la formación de una vesícula conocida como autofagosoma. Los autofagosomas se fusionan con los lisosomas, donde tiene lugar la degradación de su contenido gracias a una serie de hidrolasas que funcionan de forma óptima en el pH ácido lisosomal.
Se ha observado acumulación de autofagosomas y deficiencias lisosomales en cerebros postmortem de pacientes de Párkinson (Anglade P et al, 1997), así como en modelos animales de esta enfermedad (Dehay B et al, 2010). Sin embargo, la evidencia más clara de la relación existente entre una autofagia defectuosa y la EP la proporciona la genética. Aunque en la mayoría de los casos de EP se desconoce la causa exacta (esporádica), hay aproximadamente un 10% de casos con un origen genético claro y son debidos a mutaciones en una serie de genes, pudiendo presentar herencia autosómica dominante (LRRK2, α-sinucleína, Vps35) o autosómica recesiva (PINK1, parkina). Sorprendentemente, todas estas mutaciones se asocian con alteraciones en la ruta autofágica/lisosomal.
Mutaciones patogénicas en LRRK2, principal determinante genético de la EP, bloquean la ruta autofágica, llevando a la acumulación de material no degradado, autofagosomas y gotas lipídicas en modelos celulares y animales de EP. Estudios de nuestro laboratorio muestran que LRRK2 actúa promoviendo la liberación de calcio a través de los canales TPC2, situados en la membrana lisosomal, y provocando un incremento del pH lisosomal, lo que conllevaría una reducción de su capacidad degradativa (Gómez-Suaga P et al, 2012).
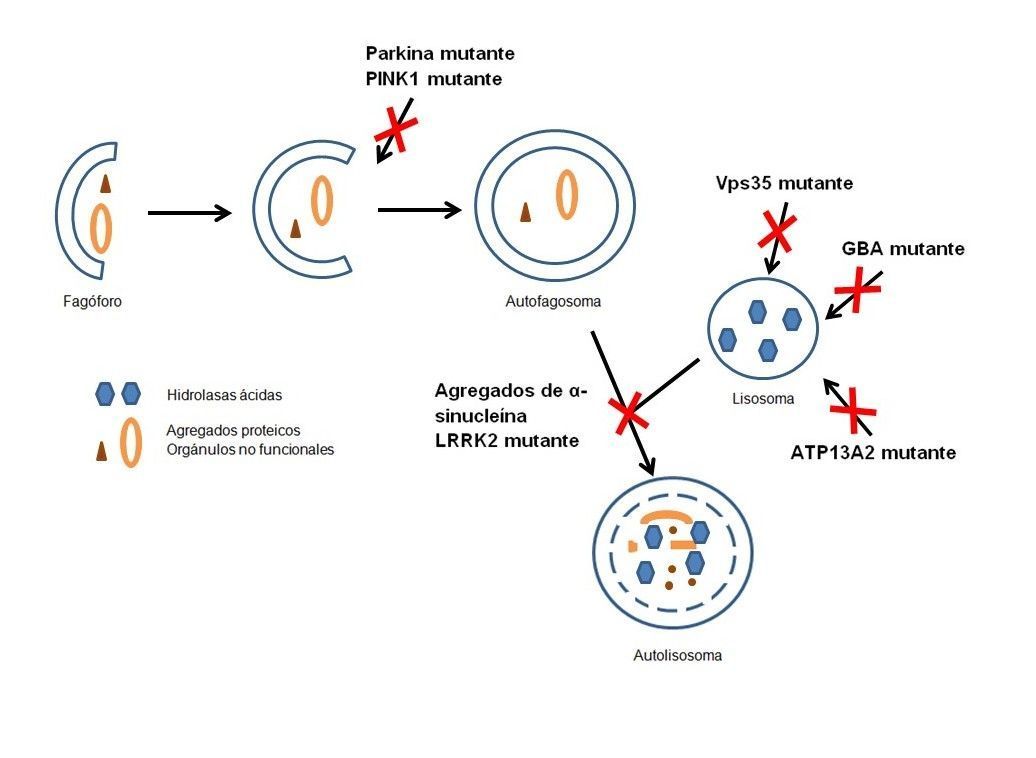
Por otra parte, la α-sinucleína es una proteína naturalmente desplegada y con gran tendencia a formar agregados, siendo de hecho uno de los principales componentes de los cuerpos de Lewy. Mutaciones que aumentan los niveles de α-sinucleína o su tendencia a la agregación promueven la formación de agregados de esta proteína, los cuales interfieren con la autofagia (Tanik SA et al, 2013).
Vps35 forma parte del retrómero, un complejo que interviene en el transporte de las hidrolasas lisosomales y un mal funcionamiento del mismo podría, por tanto, disminuir la capacidad de los lisosomas para degradar el material que reciben a través de la ruta autofágica (Follet J et al, 2014).
En cuanto a PINK1 y parkina, están involucrados en la eliminación mediante autofagia de mitocondrias, por lo que mutaciones en estos genes llevarían a la acumulación de mitocondrias dañadas, saturando la capacidad degradativa de la ruta autofágica (Wei H et al, 2015).
Además, existen al menos otros dos genes que codifican proteínas lisosomales que se han relacionado con síndromes parkinsonianos: GBA y ATP13A2 (Trinh J y Farrer M, 2013). Mutaciones en GBA, el cual codifica una enzima lisosomal implicada en el metabolismo lipídico, se asocian con mayor riesgo de desarrollar EP, lo cual podría deberse a la menor capacidad degradativa de los lisosomas como consecuencia de la acumulación lipídica en su interior. Mutaciones en ATP13A2, que codifica una ATPasa lisosomal, provocan inestabilidad de la membrana lisosomal, un mayor pH en su interior y acumulación de α-sinucleína.
Un mal funcionamiento de la autofagia resultaría, en todos los casos, en la acumulación de agregados proteicos y orgánulos dañados, que interferirían con el correcto funcionamiento de la célula, llevando finalmente a la muerte celular. Por tanto, fármacos que aumenten la autofagia podrían resultar beneficiosos para la supervivencia neuronal y retrasar la neurodegeneración asociada a la EP. Rapamicina y su análogo CCI-779, compuestos inductores de la autofagia, han mostrado efectos beneficiosos en varios modelos de EP, reduciendo la acumulación de agregados proteicos de α-sinucleína y disminuyendo la muerte neuronal (Malagelada C et al, 2010).
Además hay una serie de compuestos aprobados por la Administración de Alimentos y Medicamentos del Gobierno de Estados Unidos que actúan como inductores de la ruta autofágica y podrían ser utilizados para tratar enfermedades que impliquen alteraciones en esta ruta, aunque todavía no han sido ampliamente estudiados en modelos de EP. Entre estos compuestos se incluyen litio, carbamezipina, verapamil, calpastatina, NF449 y xestospongin B, los cuales actúan sobre diferentes moléculas y canales que intervienen en la autofagia (Sarkar S et al, 2013).
Todavía quedan muchas cuestiones por resolver para que estos compuestos se puedan implementar de forma exitosa como tratamiento frente a la EP. En primer lugar, será un desafío establecer la dosis de tratamiento adecuada para evitar la degradación excesiva de componentes celulares. Por otra parte, la mayoría de los fármacos disponibles actúan sobre otros procesos biológicos además de la autofagia, pudiendo tener efectos secundarios no deseados. Finalmente, es necesario desarrollar inductores de la autofagia dirigidos frente a tipos celulares específicos, con el objetivo de evitar efectos secundarios difíciles de controlar en otros tejidos.
Aunque queda mucho trabajo para que los compuestos moduladores de la autofagia se puedan emplear frente a la EP, los datos actuales son prometedores e indican su potencial como futura estrategia terapéutica.
Referencia:
Rivero-Ríos P, et al. Targeting the autophagy/lysosomal degradation pathway in Parkinson’s disease. Curr. Neuropharmacol. 2016; 14(3): 238-249. doi: 10.2174/1570159X13666151030103027
Bibliografía:
Anglade P, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histophathol. 1997; 12(1):25-31
Dehay B, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010; 30(37): 12535-12544. doi: 10.1523/JNEUROSCI.1920-10.2010
Follet J, et al. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic. 2014; 15(2): 230-244. doi: 10.1111/tra.12136
Gómez-Suaga P, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012; 21(3): 511-525. doi: 10.1093/hmg/ddr481
Malagelada C, et al. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010; 30(3): 1166-1175. doi: 10.1523/JNEUROSCI.3944-09.2010
Sarkar S, et al. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013; 41(5): 1103-1130. doi: 10.1042/BST20130134
Tanik SA, et al. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013; 288(21): 15194-15210. doi: 10.1074/jbc.M113.457408
Trinh J, y Farrer M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013; 9(8): 445-454. doi: 10.1038/nrneurol.2013.132
Wei H, et al. Selective removal of mitocondria via mitophagy: distinct pathways for different mitocondrial stresses. Biochim. Biophys. Acta. 2015; S0167-4889(15): 00114-00117. doi: 10.1016/j.bbamcr.2015.03.013

