
Jose Vicente Torres Pérez. School of Engineering and Materials Science. Queen Mary, University of London. Mile End Road, London, E1 4NS, United Kingdom.
Identificada la fosforilación de la serina 10 de la histona H3 como una modificación epigenética en neuronas espinales por estímulos dolorosos, y cuyo bloqueo reduce el desarrollo del dolor.
Actualmente, el tratamiento del dolor persistente sigue siendo uno de los principales retos de la medicina moderna. En el caso de las quemaduras graves, se ha conseguido mejorar la tasa de supervivencia, así como la prevención de las mismas, pero el dolor asociado a estas sigue sin tratarse eficientemente (WHO, 2008; Laycock, 2013). De hecho, el dolor prolongado por quemaduras es una de las principales fuentes de insatisfacción de los supervivientes, equiparable a la recuperación de la movilidad en sus extremidades o al aspecto físico.
La persistencia del dolor está causada, en parte, por un proceso patológico de plasticidad neuronal conocido como sensibilización central (Ji, 2003; Marchand, 2008). Este se considera un tipo de potenciación a largo plazo (LTP, de sus siglas en inglés: long-term potentiation) y se establece por la inducción de cambios de larga duración en las neuronas del sistema nervioso central, lo que genera una respuesta exagerada tras su estimulación. La sensibilización central es especialmente importante en la médula espinal superficial, el lugar principal en donde los receptores del dolor establecen conexiones sinápticas con neuronas de segundo orden (Ji, 2003; Marchand, 2008; Woolf, 2011).
En estas neuronas, la información nociva se procesa vía la activación de las MAP quinasas (MAPK: mitogen-activated protein kinases) como son ERK1/2 (extracellular signal-regulated kinases) y p38, ambas implicadas en la generación de LTP y rápidamente activadas en respuesta a diferentes estímulos nocivos (Ji, 2003). Una vez activadas, las MAPKs modifican la actividad neuronal por medio de dos tipos de mecanismos: modulando la distribución y las propiedades físicas de diferentes componentes celulares, y regulando la expresión genética (Julius, 2001; Ji, 2003). No obstante, la acción de las MAPKs en la regulación genética aún no se comprende plenamente.
Mecanismos epigenéticos, como son las modificaciones post-traduccionales de las histonas, tienen un papel clave en la regulación de la tasa de transcripción genética durante diferentes procesos, incluyendo el desarrollo y mantenimiento de la respuesta de dolor. De entre las diferentes modificaciones post-traduccionales, la fosforilación de la serina 10 de la histona H3 (pS10H3) es una modificación clásicamente considerada permisiva de la transcripción genética, regulada por MAPKs y asociada a un pequeño grupo de genes que incluye: c-fos, c-jun, c-myc y zif-268, también implicados en la respuesta al dolor (Denk, 2012; Geranton, 2012). Lo que es más importante, investigaciones previas demuestran que hay un incremento de pS10H3 en las neuronas del hipocampo (sistema nervioso central) en respuesta a diferentes tipos de estimulación, un fenómeno que se asocia con procesos de plasticidad neuronal y consolidación de la memoria (Chandramohan, 2007).
Por lo tanto, en este artículo, desarrollado en el Imperial College de Londres (Torres-Perez, 2017) postulamos que, debido a las similitudes entre los procesos de formación de la memoria (LTP) y la transmisión de la información nociva (nocicepción), pS10H3 podría estar implicada en el procesado neuronal del dolor.

Para evaluar esta hipótesis, roedores fueron sometidos a diferentes tipos de estimulación nociva, incluyendo la inducción de quemaduras graves, la inyección de neurotoxinas (capsaicina, compuesto químico responsable del escozor de los chiles, o carragenato) y la estimulación eléctrica del nervio safeno. De estos experimentos se obtuvieron tanto datos histológicos, como de concentración de proteínas (western blot) y comportamentales. Dichos experimentos cumplen con la normativa europea y la inglesa (Animals (Scientific Procedures) Act 1986 (UK) Amendment Regulations 2012 (SI 2012/3039)), y están incluidos en las directrices del Comité para la Investigación y Asuntos Éticos del IASP publicado en Pain, 16 (1983) 109- 110.
Observamos un rápido y transitorio incremento de pS10H3 en el núcleo de una sub-población de neuronas de la médula espinal superficial después de dichos modelos de dolor. De dichas neuronas, al menos una gran parte de ella corresponden a neuronas de segundo orden, puesto que reciben conexiones directas de los receptores del dolor (las neuronas de primer orden).
Además, estudiando los niveles de expresión conjunta demostramos que, al igual que las neuronas del hipocampo (Chandramohan, 2007), este marcaje epigenético está controlado por las MAPKs ERK1/2 y regula la subsiguiente transcripción del gen c-fos (figura 1). Adicionalmente, con estudios farmacológicos usando bloqueadores específicos comprobamos que pS10H3 es dependiente de NMDA (N-metil-D-aspartato), un componente fundamental para el establecimiento de plasticidad neuronal y la memoria (Ji, 2003).
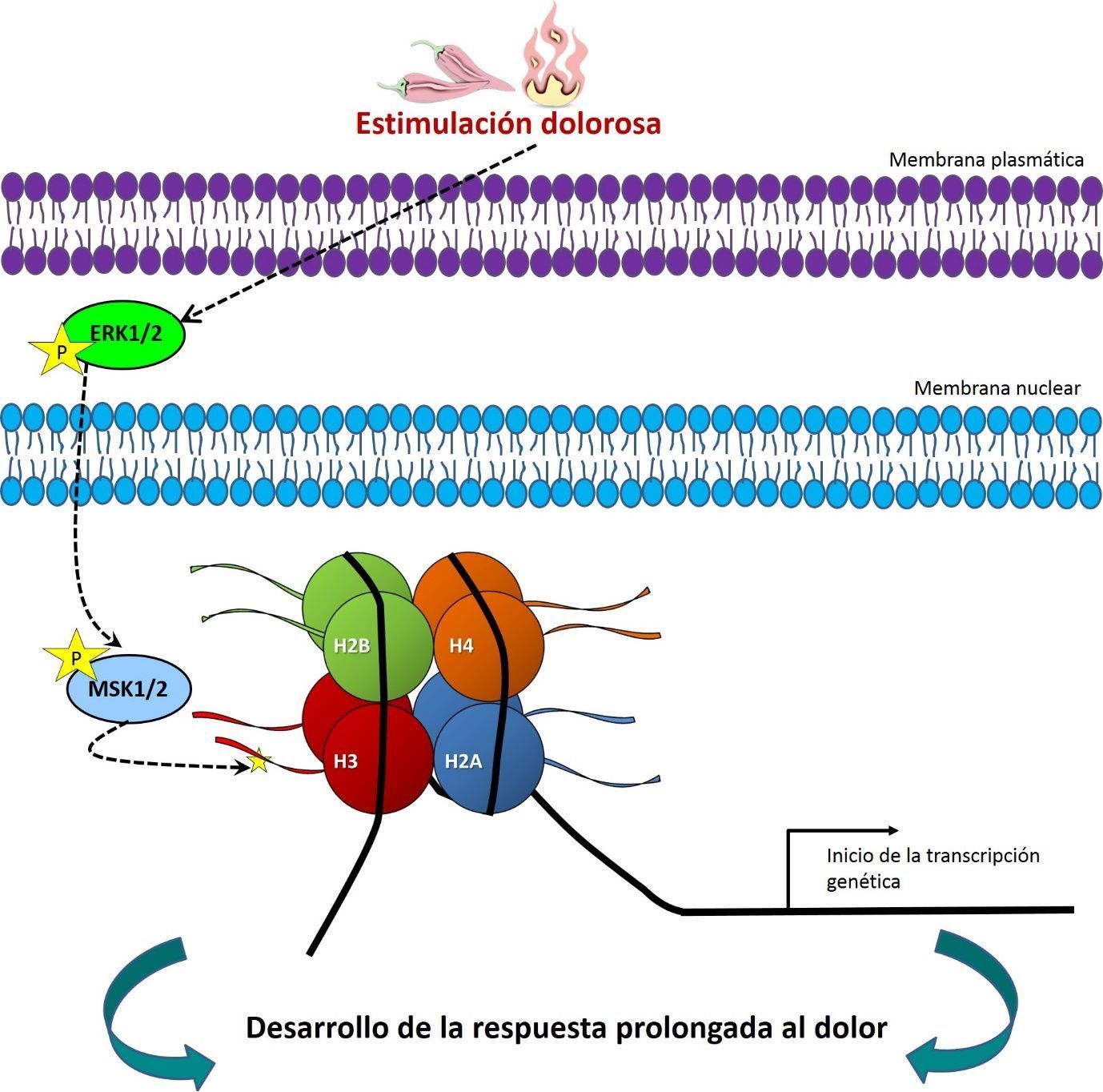
Estudios farmacológicos también demostraron que MSK1/2 (mitogen- and stress-activated protein kinases), quinasas que a si mismo son activadas por ERK1/2, son las principales enzimas encargadas de escribir pS10H3 en estas neuronas (figura 1). Consecuentemente, se estudiaron las diferencias comportamentales entre un grupo de ratones normales y otros modificados genéticamente para no expresar estas quinasas (MSK1/2 -/-). Estos experimentos conductuales demostraron que la ausencia de MSK1/2 bloquea el desarrollo de hipersensibilidad al calor que normalmente acompaña una estimulación dolorosa.
Por todo esto, concluimos que pS10H3 es un evento clave que controla el procesado genético en una importante población de neuronas espinales en respuesta a inflamación y/o daño de tejidos (figura 1). Futuros estudios usando a pS10H3 como diana de bloqueo o para ChIp-Seq (immunoprecipitación de cromatina combinada con técnicas de secuenciación) ayudaran a entender con más detalle los eventos moleculares implicados en el desarrollo y establecimiento del dolor persistente. Por tanto, este descubrimiento podría ayudar a desarrollar nuevas estrategias analgésicas para combatir más eficientemente el dolor permanente, por ejemplo, el asociado a las quemaduras graves.
Referencia:
Torres-Perez JV, et al. Phosphorylated Histone 3 at Serine 10 Identifies Activated Spinal Neurons and Contributes to the Development of Tissue Injury-Associated Pain. Sci Rep. 2017 Jan 25;7:41221. doi: 10.1038/srep41221.
Bibliografía:
Chandramohan Y, et al. Novelty stress induces phospho-acetylation of histone H3 in rat dentate gyrus granule neurons through coincident signalling via the N-methyl-D-aspartate receptor and the glucocorticoid receptor: relevance for c-fos induction. J Neurochem. 2007 May;101(3):815-28. doi: 10.1111/j.1471-4159.2006.04396.x
Denk F, et al. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012 Feb 9;73(3):435-44. doi: 10.1016/j.neuron.2012.01.012.
Geranton SM. Targeting epigenetic mechanisms for pain relief. Curr Opin Pharmacol. 2012 Feb;12(1):35-41. doi: 10.1016/j.coph.2011.10.012.
Ji RR, et al. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003 Dec;26(12):696-705. doi: 10.1016/j.tins.2003.09.017
Julius D, et al. Molecular mechanisms of nociception. Nature. 2001 Sep 13;413(6852):203-10. doi: 10.1038/35093019
Laycock H, et al. Peripheral mechanisms of burn injury-associated pain. Eur J Pharmacol. 2013 Sep 15;716(1-3):169-78. doi: 10.1016/j.ejphar.2013.01.071.
Marchand S. The physiology of pain mechanisms: from the periphery to the brain. Rheum Dis Clin North Am. 2008 May;34(2):285-309. doi: 10.1016/j.rdc.2008.04.003.
WHO (World Health Organization). A WHO plan for burn prevention and care. 2008. URL: http://apps.who.int/iris/bitstream/10665/97852/1/9789241596299_eng.pdf [08-02-2017]
Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011 Mar;152(3 Suppl):S2-15. doi: 10.1016/j.pain.2010.09.030.

