
Janire Mingo1, Sandra Luna1, Teresa Fernández-Acero2, Isabel Rodríguez-Escudero2
1 Instituto de Investigación Sanitaria Biocruces, Barakaldo, Bizkaia, España.
2 Departamento de Microbiología y Parasitología, Facultad de Farmacia, Universidad Complutense de Madrid, e Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid, España.
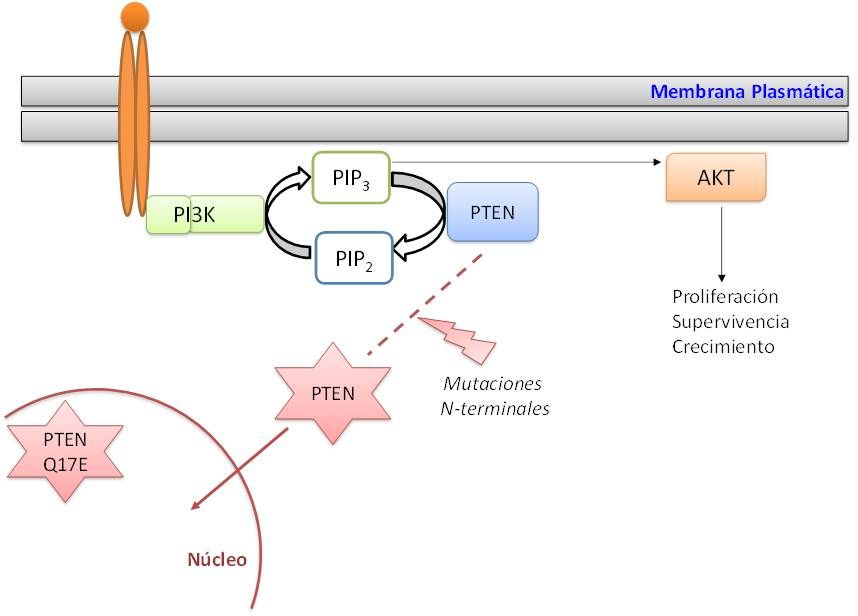
PTEN es uno de los genes supresores tumorales mutados con mayor frecuencia en cánceres humanos. La proteína a la que da lugar, PTEN, ejerce funciones esenciales tanto en el citoplasma como en el núcleo celular y forma parte de la ruta de señalización de la fosfatidilinositol 3-quinasa (PI3K), que genera en la membrana plasmática fosfatidil-inositol 3,4,5-trifosfato (PIP3) en respuesta a estímulos extracelulares, y cuyo principal efector es la proteína quinasa B (PKB o Akt). Esta ruta se encuentra hiperactivada a distintos niveles en muchos tumores en humanos. Aunque PTEN pertenece a la familia de las fosfatasas de tirosina, su principal acción como supresor de tumores se debe a su actividad catalítica fosfatasa de lípidos sobre PIP3, razón por la cual actúa contrarrestando la activación de la ruta PI3K/Akt.
La estructura de PTEN consta de un extremo N-terminal seguido por el dominio fosfatasa catalítico, un dominio C2 de unión a membrana y un extremo C-terminal con un motivo de unión a proteínas PDZ.
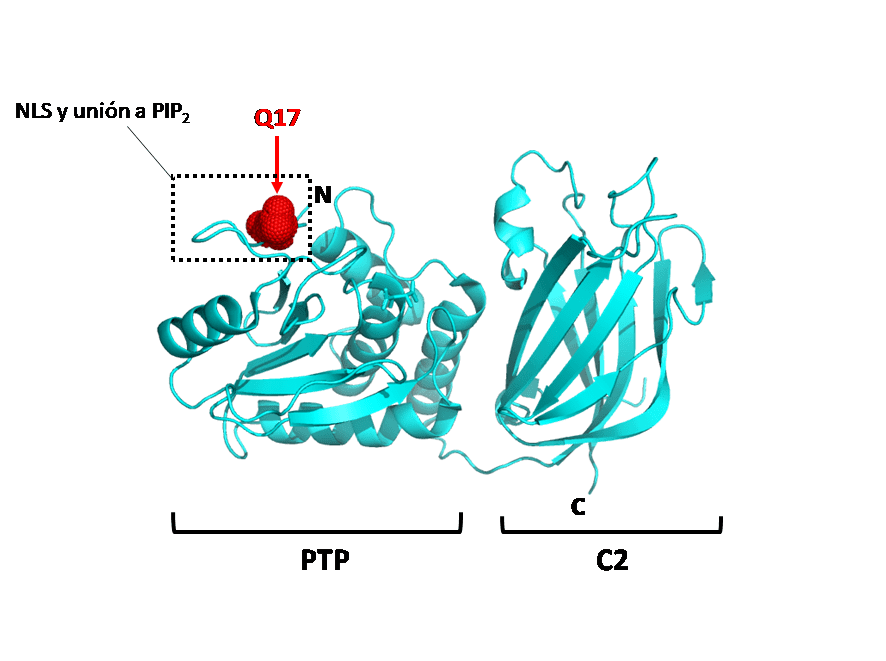
La región N-terminal de PTEN es importante para el control de su localización subcelular y su función. Contiene una señal de localización nuclear (NLS) y un motivo de unión al fosfatidilinositol 4,5-bifosfato (PIP2) implicado en el direccionamiento a la membrana plasmática y activación de la enzima. La interacción de PTEN con la membrana plasmática es esencial para que desfosforile el PIP3, evitando así la proliferación y crecimiento celular excesivos. De hecho, se han encontrado mutaciones somáticas asociadas a tumores en el extremo N-terminal de PTEN que reducen su capacidad para unirse a la membrana plasmática.
En este trabajo, hemos caracterizado funcionalmente la variante Q17E (Gln17Glu), en la que la glutamina de la posición 17 de PTEN es sustituida por un glutámico, la cual ha sido descrita tanto en pacientes con PHTS como en pacientes con ASD. El aminoácido Gln17 se localiza en los motivos de localización subcelular en la región N-terminal de PTEN, lo que sugiere su posible relevancia para la función de la proteína.
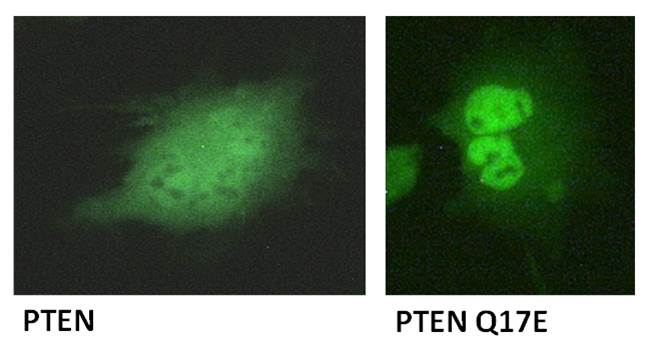
La variante Q17E muestra una actividad fosfatasa ligeramente reducida y se acumula en el núcleo celular
Hemos analizado, tanto en un modelo experimental in vivo basado en expresión heteróloga en la levadura Saccharomyces cerevisiae como en células de mamíferos, la funcionalidad de la variante Q17E. A diferencia de otras variantes patológicas de PTEN, que afectan fuertemente a la catálisis o a la estabilidad de la proteína, la variante Q17E mostró una actividad fosfatasa ligeramente disminuida y una estabilidad proteica normal en las células. Dicha variante no se ha encontrado hasta la fecha en bases de datos de mutaciones somáticas de cáncer. Además, mediante inmunofluorescencia y técnicas de microscopía, nos propusimos determinar la localización subcelular de la variante Q17 de PTEN. Pudimos observar cómo PTEN Q17E se acumula en el núcleo, a diferencia de PTEN que es principalmente citoplasmático.
Las variantes hereditarias del extremo N-terminal de PTEN causan acumulación nuclear de PTEN
Adicionalmente, estudiamos la localización subcelular de un grupo de variantes N-terminales de PTEN (22 variantes, comprendidas entre los residuos 13-32) encontradas en la línea germinal de pacientes con PHTS y ASD.
Los resultados revelaron una mayor acumulación nuclear de las variantes asociadas a enfermedad y portadoras de mutaciones entre los residuos 17-26. Asimismo, el análisis funcional in vivo de la actividad fosfatasa sobre PIP3 en levadura puso de manifiesto la pérdida de función parcial o total para la mayoría de estas variantes, sugiriendo una relación entre el déficit en la actividad catalítica de PTEN en las células y su acumulación nuclear.
Nuestros resultados sugieren que la acumulación nuclear de las variantes N-terminales de PTEN, podría estar ligada a la pérdida de su actividad fosfatasa sobre PIP3, lo que podría ser un rasgo característico de las mutaciones de PTEN encontradas en estos síndromes. De esta forma, la retención en el núcleo de estas versiones mutadas en la región N-terminal (como la citada PTEN Q17E) podría ser uno de los mecanismos que impide a la proteína ejercer su actividad fosfatasa sobre el PIP3 en la proximidad de la membrana plasmática.
En conjunto, nuestros resultados indican que la localización subcelular y la actividad fosfatasa de PTEN dependen de la integridad de su extremo N-terminal. Por tanto, la posibilidad de interferir en su función mediante el control de su localización es una opción a tener en cuenta para la prevención o tratamiento de enfermedades asociadas a PTEN.
Nuestros resultados contribuyen a entender por qué ciertas mutaciones de PTEN que afectan a su localización contribuyen a la patología de síndromes de tipo PHTS y ASD, aunque quedan abiertos aún muchos interrogantes respecto a los mecanismos moleculares implicados.
Referencia: Mingo J, Rodríguez-Escudero I, et al. A pathogenic role for germline PTEN variants which accumulate into the nucleus. European Journal of Human Genetics. 2018; 10.1038/s41431-018-0155-x.

