Jose Miguel Laffita Mesa1,2
1 Neuroscience (CNS), Neuro Svenningsson, J5:20 Bioclinicum, Karolinska Universitetssjukhuset 171 76 Stockholm, Sweden.
2 Department NVS, Division of Neurogeriatrics, Karolinska Institutet. Unit for Hereditary Dementias, Theme Aging | Karolinska University Hospital-Solna, Stockholm, Sweden.
El gen ataxin-2 (ATXN2) tiene una secuencia repetitiva de tripletes de CAG que varía desde 13 a 31 unidades. Al sobrepasar ciertos umbrales se asocia o causa varias enfermedades neurodegenerativas en humanos, como ataxia espinocerebelosa tipo 2 (SCA2), esclerosis lateral amiotrófica (ELA), enfermedad de Parkinson (EP) y otras. Salvo algunos SNPs asociados con efecto sobre la longevidad y fenotipos metabólicos, la longitud anormal de este triplete es, de lejos, la única alteración genética de ATXN2 conocida en enfermedades neurológicas. Es además la más importante fisiológicamente, dado que su alteración causa pérdida o ganancia de funciones tóxicas de ataxina-2 que desencadenan la neurodegeneración.
Varios estudios han demostrado la interacción entre ATXN2 y C9ORF72, por lo que durante el estudio de la metilación del gen C9ORF72 en muestras de ADN del repositorio Coriell (Camden, NJ, USA), secuenciamos el gen ATXN2 para el genotipaje de los SNP rs695871, rs695872 y rs7969300. Haciendo esto, identificamos, por serendipia, la duplicación de la secuencia c.109_117delinsCGGAGCGGG (ref seq. NM_002973). Esta alteración se localiza en la región digénica ATXN2–Sense/Antisense, y causa la duplicación del motivo Ser-Gly-Arg. Para el gen ATXN2-S la duplicación está en la región promotora/Exon-1, y para ATXN2-AS a 60 pares de bases del final del intrón-1 (Fig. 1a).
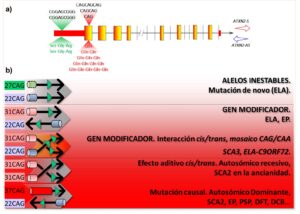
La alteración fue encontrada en casos de ELA con la expansión de nucleótidos GGGGCC en C9ORF72 (ELA-C9ORF72), en una familia sueca con SCA3 (otra enfermedad causada por repeticiones de CAG, pero en el gen ATXN3) y en la enfermedad de Parkinson. Por el contrario, solo estuvo presente en 0.48% de los 823 controles de varias poblaciones. El escrutinio de las bases de datos genómicos: gnomAD, ExAC, SNPdB y el proyecto 1000 Genomas reveló ausencia de la mutación (Laffita-Mesa et al., 2020).
La duplicación modifica la edad de debut de la SCA3, lo cual surge de una posible interacción alélica con expansiones intermedias del gen ATXN2. El mismo efecto fue confirmado en casos ELA-C9ORF72. No obstante, aquí la edad de debut de los portadores de la duplicación fue menor que en casos ELA-C9ORF72 y con expansiones intermedias en ATXN2. En uno de los casos con la duplicación comprobamos un fenómeno de mosaico para el segmento de CAG. En los casos con EP sin mutaciones no se verificó el mismo efecto, confirmando un auténtico efecto de gen modificador para esta alteración de ATXN2.
Dado que existían antecedentes contradictorios acerca de la inclusión del segmento promotor en el transcripto principal, realizamos estudios de expresión mediante PCR digital de gotas. Se comprobó la inclusión de la duplicación en el transcripto principal de ATXN2 y observamos un aumento significativo en los niveles de ATXN2 en los casos con la duplicación.
La confirmación del efecto modificador en estas dos enfermedades cuyos genes son sensibles a las pérdida o ganancia de función de ATXN2, así como la exclusión de otros modificadores en ambas cohortes, validan fuertemente la hipótesis de la duplicación como modificadora del fenotipo. Las expansiones en C9ORF72 son, pero por mucho, la alteración genética más frecuente asociada a ELA y demencia frontotemporal. Al igual, la SCA3 es la ataxia espinocerebelosa más frecuente a nivel global y se necesitan terapias para estas enfermedades humanas huérfanas de tratamiento.
Varios estudios descartaron otras alteraciones genéticas en ATXN2 (Pulst et al., 1996, Imbert et al., 1996, Sanpei et al., 1996, Figueroa et al., 2009). Incluso, Wang et al., 2015 usaron análisis de ligamiento y secuenciación exómica y concluyeron que el único defecto genético es el triplete repetitivo de CAG, como la explicación de la variabilidad fenotípica en SCA2 parkinsoniana. Por tanto, nuestro estudio es el primero en demostrar un defecto genético distinto al triplete de CAG en ATXN2.
Este estudio realizado en el Instituto Karolinska añade un nuevo mecanismo genético de este gen, el cual es un modificador o causa de varias enfermedades. La complejidad genética de ATXN2 es tal que al menos se suponen cuatro mecanismos distintos influyendo o causando enfermedades neurológicas, siendo el nuestro un nuevo mecanismo que pudiera, a la vez, estar presente en los otros (Fig. 1b).
Estos hallazgos ayudarán a entender las fuentes de la variabilidad biológica encontrada en casos con ataxia y otras enfermedades donde ATXN2 está involucrada directa e indirectamente. A su vez permitirá proveer pronósticos certeros a los afectados, así como a sus familiares. También posibilitará estratificar casos de acuerdo con modificadores genéticos en ensayos clínicos como la terapia con oligonucleótidos antisentido que pronto será ensayada en varias cohortes con SCA2 aquí y en varios países. Y desde una visión más científica, a largo plazo, ayudará a entender la función biológica de ATXN2.
Referencia: Laffita-Mesa JM et al. A novel duplication in ATXN2 as modifier for spinocerebellar ataxia 3 (SCA3) and C9ORF72-ALS. Movement disorders. 2020. doi:10.1002/mds.28334
Bibliografía
Figueroa K et al. Genetic Variance in the Spinocerebellar Ataxia Type 2 (ATXN2) Gene in Children with Severe Early Onset Obesity. PLoS One. 2009; 4(12).
Imbert G et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14(3):285-91.
Pulst SM et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14(3):269-76.
Pulst SM. The complex structure of ATXN2 genetic variation. Neurol Genet. 2018 6;4(6):e299.
Sanpei K et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14(3):277-84.
Wang C et al. Linkage analysis and whole-exome sequencing exclude extra mutations responsible for the parkinsonian phenotype of spinocerebellar ataxia-2. Neurobiol Aging. 2015;36(1):545.e1-7.
Si te ha gustado esta noticia y quieres aprender más sobre Genética en Medicina, te interesan nuestros cursos y formación universitaria.



