
Rocío Tejero, Mario López-Manzaneda, Saravanan Arumugam, Lucía Tabares
Departamento de Fisiología Médica y Biofísica. Facultad de Medicina. Universidad de Sevilla
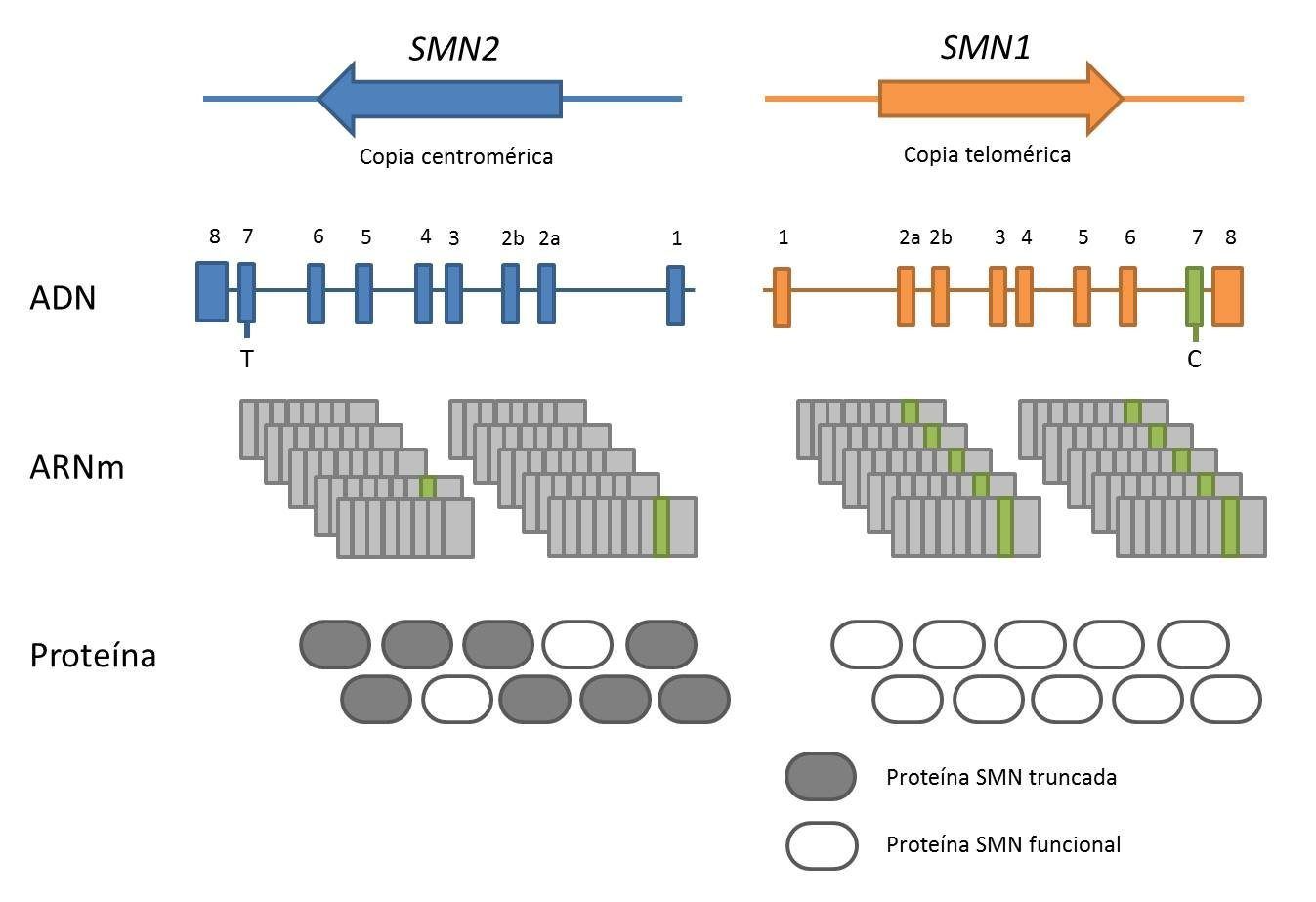
La Atrofia Muscular Espinal (AME) es una enfermedad neurodegenerativa, monogénica autosómica recesiva, caracterizada por pérdida de motoneuronas inferiores, debilidad muscular y parálisis. En la actualidad es la enfermedad genética que causa más mortalidad infantil y la segunda enfermedad autosómica recesiva más frecuente, tras la fibrosis quística, con una incidencia de 1:10.000 nacidos vivos y una frecuencia de portadores entre 1:40-1:60 (Crawford and Pardo, 1996). La AME está causada por la pérdida o mutación en homocigosis del gen de Supervivencia de Motoneuronas 1 (SMN1), gen en posición telomérica (Lefebvre et al., 1995). En humanos existe un duplicado de SMN1 de posición centromérica, el gen SMN2, cuyo número de copias varía entre la población. En ausencia de SMN1, la severidad de la enfermedad correlaciona de manera inversa con el número de copias de SMN2.
SMN1 y SMN2 poseen una secuencia génica homóloga, con una identidad de un 99,9%. Se diferencian en cinco pares de bases, siendo la única diferencia funcionalmente relevante la transición citosina-timina (C-T) en el exón 7. Esta transición hace que mientras que SMN1 produce proteína de Supervivencia de Motoneuronas (SMN) completa, SMN2 produce mayoritariamente proteína SMN truncada (SMNΔ7), la cual es inestable y rápidamente degradada. SMN es una proteína de 294 aminoácidos, ubicua, que se encuentra en el núcleo y en el citosol de las células. Su función mejor caracterizada es la participación en el ensamblaje de las ribonucleoproteínas nucleares pequeñas (snRNP) y en la maduración del ARNm en el espliceosoma.
En distintos modelos animales de la enfermedad la unión neuromuscular presenta alteraciones estructurales y funcionales, estando la liberación de neurotransmisor muy disminuida (Kong et al., 2009, Ruiz et al., 2010, Torres-Benito et al., 2011). No obstante, no todos los contingentes de neuronas motoras y músculos se encuentran afectados en la misma medida, estando más afectados los músculos axiales y proximales que los distales.
En nuestro trabajo (Tejero et al., 2016) hemos investigado el origen del déficit de la liberación de neurotransmisor en los terminales nerviosos motores y las bases moleculares de la afectación selectiva de unos grupos musculares sobre otros. Para ello, comparamos las propiedades moleculares y funcionales de los terminales nerviosos en músculos con diferente grado de afectación en el modelo de ratón SMNΔ7.
Los resultados revelan que los niveles de expresión de la proteínas sinápticas sinaptotagmina-2 (Syt2) y la proteína de la vesícula sináptica 2 (SV2) B están muy disminuidos en los terminales nerviosos motores AME, en comparación con los controles, mientras que los de otras proteínas sinápticas, como sintaxina-1B (Stx1B) y sinaptotagmina-7 (Syt7), no están alterados.
También hemos hallado que durante el desarrollo postnatal en los terminales motores se produce un cambio en la expresión de las isoformas 1 y 2 de sinaptotagmina, de forma que Syt1 se regula a la baja y Syt2 al alza. En los terminales deficientes en SMN de los músculos más afectados, la disminución en los niveles de Syt2 junto con los bajos niveles de Syt1 hacen a la sinapsis altamente vulnerable. Además, estos terminales deficientes presentan una reducción en la densidad de los canales de Ca2+ dependientes de voltaje tipo P/Q, lo que contribuye también a la disminución en la liberación de neurotransmisor.
En consonancia con los anteriores resultados, el análisis funcional de la neurotransmisión en las sinapsis neuromusculares más afectadas reveló una gran reducción en la liberación evocada, una alteración de la plasticidad a corto plazo, una reducción en la probabilidad de liberación vesicular y la incapacidad de los terminales para modular normalmente el número de sitios de liberación activos.
En resumen, estos datos sugieren que la gran reducción de dos proteínas muy relevantes en la sinapsis, Syt2 y SV2B, da lugar al déficit funcional y que la regulación fisiológica de Syt1 desempeña un papel importante en la vulnerabilidad muscular selectiva en AME.
Referencia: Tejero R, et al. Synaptotagmin-2, and -1, linked to neurotransmission impairment and vulnerability in Spinal Muscular Atrophy. Hum Mol Genet. 2016 Aug 29. Doi: 10.1093/hmg/ddw297
Bibliografía:
Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996 Apr;3(2):97-110. Doi: 10.1006/nbdi.1996.0010
Kong L, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009 Jan 21;29(3):842-51. doi: 10.1523/JNEUROSCI.4434-08.2009.
Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995 Jan 13;80(1):155-65. Doi: 10.1016/0092-8674(95)90460-3
Ruiz R, et al. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci. 2010 Jan 20;30(3):849-57. doi: 10.1523/JNEUROSCI.4496-09.2010.
Torres-Benito L, et al. SMN requirement for synaptic vesicle, active zone and microtubule postnatal organization in motor nerve terminals. PLoS One. 2011;6(10):e26164. doi: 10.1371/journal.pone.0026164.

