
Nerea Moreno, Estefanía Cerro-Herreros, María Sabater-Arcis, Juan M. Fernández-Costa, Manuel Pérez-Alonso, Beatriz Llamusi y Rubén Artero
Estructura de Investigación Interdisciplinar en Biotecnología y Biomedicina de la Universitat de València
Instituto de Investigación Sanitaria INCLIVA
Investigadores del grupo de Genómica Traslacional de la Universidad de Valencia-INCLIVA describen un posible tratamiento para la distrofia miotónica tipo 1 a partir del silenciamiento de miARNs. El trabajo ha sido publicado en la revista científica Nature Communications y demuestra el potencial terapéutico del aumento de MBNL1/2 mediante terapia génica.
La distrofia miotónica tipo 1 es una enfermedad genética autosómica dominante causada por la expansión patológica de CTG en el 3’ UTR del gen DMPK. Estas repeticiones en los transcritos provocan el secuestro de proteínas de unión al ARN como MBNL1/2. El secuestro de MBNL1 desencadena alteraciones en el metabolismo del ARN, particularmente en el control del corte y empalme alternativo de transcritos musculares, cardíacos y neuronales, lo que se define como espliceopatía.
Clínicamente la DM1 se considera un trastorno multisistémico cuya gravedad está correlacionada con el tamaño de expansiones. Entre sus síntomas destaca la miotonía muscular, definitoria de la enfermedad. La miotonía se define como la incapacidad de relajar el músculo tras una contracción voluntaria. Además, los pacientes padecen debilidad muscular y atrofia, tanto en músculo liso como esquelético. La enfermedad también cursa con disfunciones del sistema nervioso central (SNC).
A pesar del esfuerzo en hallar una estrategia eficaz para la DM1 ninguna ha llegado todavía a la clínica. Las dos estrategias terapéuticas principales que se han abordado en los últimos años se centran en la degradación de los transcritos de DMPK mutantes o en evitar el secuestro de MBNL1/2 (Thornton et al. 2017). Sin embargo, el trabajo realizado por el grupo de Genómica traslacional se basa en una estrategia novedosa que consiste en la modulación terapéutica de la expresión génica endógena de MBNL1/2.
En un estudio anterior de prueba de concepto con Drosophila se demostró que la regulación al alza de MBNL es una terapia prometedora para la DM1 y que esto se podía conseguir con el silenciamiento de miARNs específicos en moscas modelo de la enfermedad (Cerro-Herreros et al. 2016). Los microARNs son ARNs pequeños no codificantes que reprimen la expresión del ARN mensajero (mARN) mediante su unión al 3’UTR del mismo. En el presente trabajo los autores trasladaron esta estrategia terapéutica a modelos mamíferos de la enfermedad.
Para ello los investigadores comenzaron con un rastreo en el que identificaron en cultivos celulares HeLa algunos microRNAs como potenciales represores de MBNL1/2, entre los que destacaron dos microARNs como dianas terapéuticas en DM1: miR-23b y miR-218. Para su inhibición diseñaron oligonucleótidos antisentido de secuencia complementaria a miR-23b y miR-218 llamados AntagomiRs. Estos oligonucleótidos incluían algunas modificaciones químicas que facilitaban su entrada en la célula y mejoraban la eficiencia y estabilidad. Los AntagomiRs se unen al microARN impidiendo el reconocimiento de sus transcritos diana e impiden el bloqueo que ejercen estos sobre la expresión génica (Figura 1).
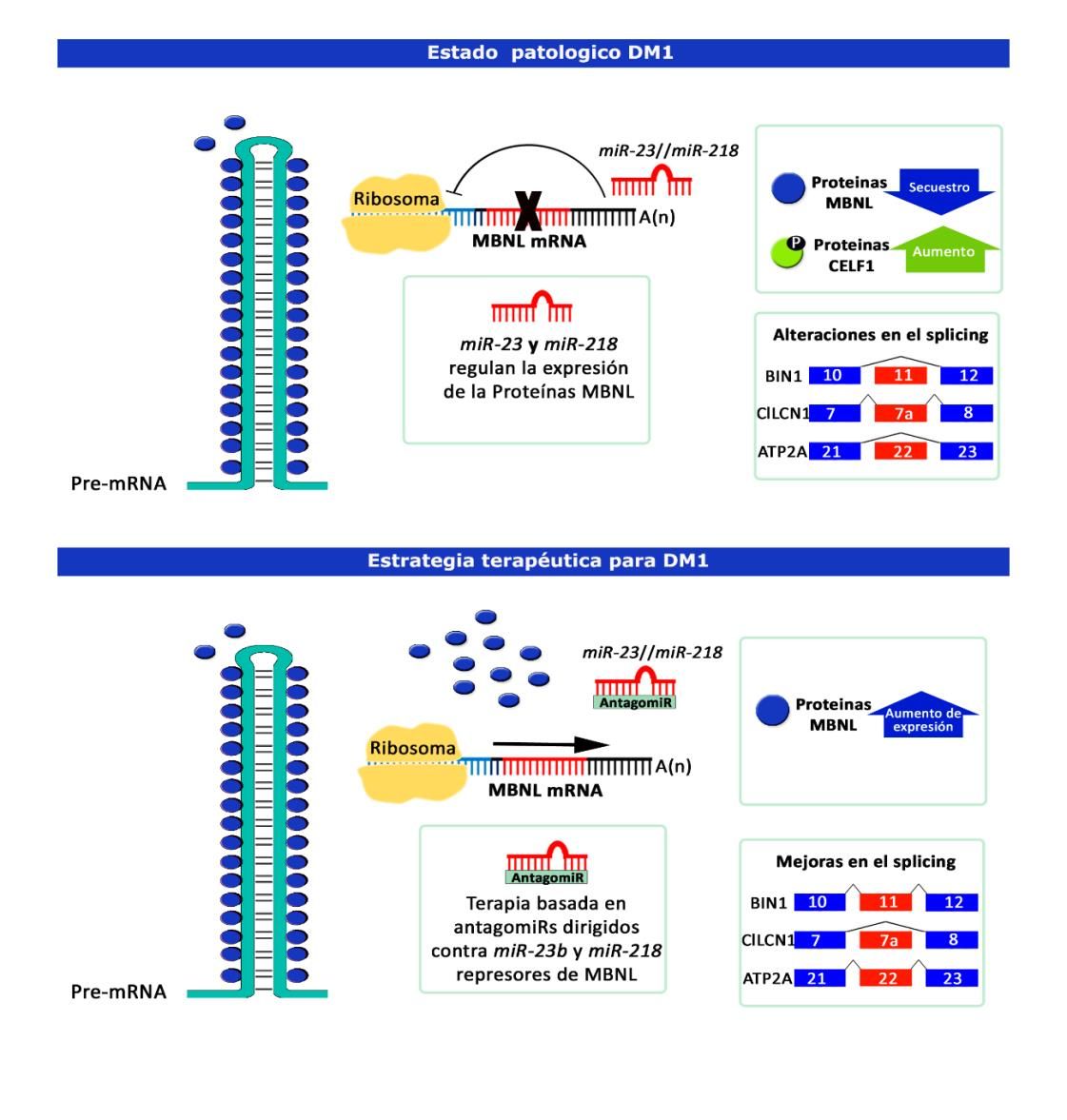
Dado que los microARNs están implicados en la regulación de la expresión génica tanto a nivel de estabilidad del mARN como a nivel de su traducción a proteína, procedieron a determinar el efecto que provocaba el silenciamiento de los microARN a este nivel. Se concluyó que el tratamiento daba lugar a un aumento significativo de la expresión del mensajero y las proteínas de MBNL en mioblastos de pacientes DM1 y que éste rescataba la espliceopatía típica de la patología.
Con la finalidad de investigar la actividad in vivo que podían tener los AntagomiRs decidieron administrar el tratamiento a ratones modelo de la enfermedad (HSALR). Primeramente se comprobaron dos circunstancias claves para el trabajo. Por un lado, que los miARNs se expresaran en los tejidos de interés y por otro lado, que los AntagomiRs llegaban a estos tejidos (gastronemio y cuádriceps). Como consecuencia de la administración sistémica de los AntagomiRs se observó la disminución de la expresión de los microARN miR-23b y miR-218 y el aumento de los niveles de Mbnl1 y Mbnl2 a nivel de transcrito y de proteína en los tejidos elegidos para el estudio.
Además el tratamiento produjo el rescate de la histopatología muscular, ya que los AntagomiRs disminuyeron el número de núcleos centrales en fibras musculares, producto del intento del musculo por tratar de regenerarse y que es una característica típica de la enfermedad. Por otro lado, también reducía la miotonía en los ratones y aumentaba la fuerza muscular que tenían antes del tratamiento.
Durante el trabajo también se confirmó la expresión de miR-23b y miR-218 en otros tejidos importantes para la patología como corazón y SNC, por lo que es de esperar que el aumento en las proteínas MBNL1 y MBNL2 por la administración sistémica de los AntagomiRs pueda rescatar fenotipos en estos tejidos.
A pesar del hecho de que estos microARNs tienen como diana natural otros genes y su silenciamiento podría provocar otros efectos no deseados, este silenciamiento en condiciones patológicas supera a las posibles alteraciones deletéreas, ya que los análisis de necropsia visual y bioquímica sanguínea de los ratones tratados tanto a corto como a largo plazo reveló que no existían efectos nocivos.
Todos estos resultados en conjunto demuestran que el silenciamiento de miR-23b y miR-218 produce un aumento de MBNL1/2 que se traduce en el rescate de las alteraciones moleculares, histológicas y funcionales tanto en mioblastos de pacientes como en ratones DM1. También confirman el potencial de los oligonucleotídicos para desreprimir específicamente la expresión de las proteínas MBNL1 y MBNL2 como enfoque terapéutico para la distrofia miotónica tipo 1.
Investigación original:
Cerro-Herreros E, et al. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat Commun. 2018 Jun 26;9(1):2482. doi: http://dx.doi.org/10.1038/s41467-018-04892-4
Bibliografía:
Cerro-Herreros E, et al. Derepressing muscleblind expression by miRNA sponges ameliorates myotonic dystrophy-like phenotypes in Drosophila. Sci Rep. 2016 Nov 2;6:36230. doi: 10.1038/srep36230.
Thornton CA, et al. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev. 2017 Jun;44:135-140. doi: http://dx.doi.org710.1016/j.gde.2017.03.007

