
Ines Garcia-Consuegra, PhD1,2,3, Alberto Blázquez, BS1,2,3, Juan Carlos Rubio, PhD1,2,3, Joaquín Arenas, PhD1,2, Alfonsina Ballester-Lopez, MS4, Daniëlle Coenen, BS4,5, Adrián González-Quintana, BS1,2, Antoni L. Andreu, MD, PhD6, Tomàs Pinós, PhD3,7, Jaume Coll-Cantí, MD, PhD4,8 Alejandro Lucia, MD, PhD2,9, Gisela Nogales-Gadea, PhD4#, Miguel A. Martín, PhD 1,2,3#
1Laboratorio de enfermedades mitocondriales y neuromusculares. Hospital 12 de Octubre, Madrid.
2Instituto de Investigación Hospital 12 de Octubre (i+12), Madrid.
3Centro de Investigación Biomédica en Red para las Enfermedades Raras (CIBERER), Instituto de Salud Carlos III.
4Translational Research Laboratory in Neuromuscular Diseases, Department of Neurosciences, Institut d’Investigació en Ciències de la Salut Germans Trias i Pujol i Campus Can Ruti, Universitat Autònoma de Barcelona, Badalona.
5Maastricht University, Holanda.
6Hospital Universitari de Bellvitge, Hospitalet del Llobregat.
7Institut de Recerca Val d’Hebron, Barcelona.
8Unidad Neuromuscular, Hospital Universitari Germans Trias i Pujol, Badalona.
9Universidad Europea, Madrid.
#Igual contribución en este trabajo
La enfermedad de McArdle es un trastorno metabólico caracterizado por la intolerancia al ejercicio físico. Los pacientes no pueden metabolizar el glucógeno muscular debido a la deficiencia en la isoforma muscular de la enzima glucógeno fosforilasa o miofosforilasa (Nogales-Gadea G. et al., 2016). Debido a esta deficiencia, los pacientes presentan fatigabilidad, dolor muscular, dificultad o incapacidad para realizar tareas simples de la vida diaria, y en los casos más graves, los pacientes pueden presentar debilidad crónica, que en algunas ocasiones les lleva a requerir soporte para su movilidad (Lucia A. et al., 2012).
Es una enfermedad genética de herencia autosómica recesiva, causada por mutaciones en el gen PYGM (Nogales-Gadea G. et al., 2016). Hasta el momento se han descrito más de 140 mutaciones diferentes que han sido reportadas como causa de esta patología. El incremento en la frecuencia de nuevos casos descritos por nuestro grupo y por otros, sugiere que aún un buen número de pacientes permanece sin diagnosticar (Lucia A. et al., 2012; Nogales-Gadea G. et al., 2015).
Hasta el momento, el diagnóstico de los pacientes se llevaba a cabo mediante una extracción sanguínea para estudiar las posibles mutaciones más frecuentes en el ADN. Además, se realizaba una biopsia muscular en aquellos casos en los que no se había detectado ninguna mutación con el método anterior o los cambios encontrados eran de significado incierto. A través del análisis de ARN muscular se proporcionaba diagnóstico a los casos más complicados, y en algunas ocasiones se evidenciaba la patogenicidad de los cambios genéticos encontrados (García-Consuegra I. et al., 2009).
En este estudio presentamos un nuevo protocolo de diagnóstico basado en el estudio de ARN de la miofosforilasa en células mononucleares de sangre periférica (abreviado PBMCs, del inglés, peripheral blood mononuclear cells). El objetivo es intentar diagnosticar más pacientes de McArdle y caracterizar correctamente las mutaciones, en una muestra menos invasiva que la biopsia muscular, como es la extracción de sangre. Para llevar a cabo este estudio, analizamos el ARN de la miofosforilasa en el músculo de 14 individuos (7 controles y 7 pacientes), y lo comparamos con el ARN de la miofosforilasa obtenido de PBMCs (12 pacientes y 14 controles), para ver si ambas técnicas eran igual de eficaces a la hora de detectar mutaciones.
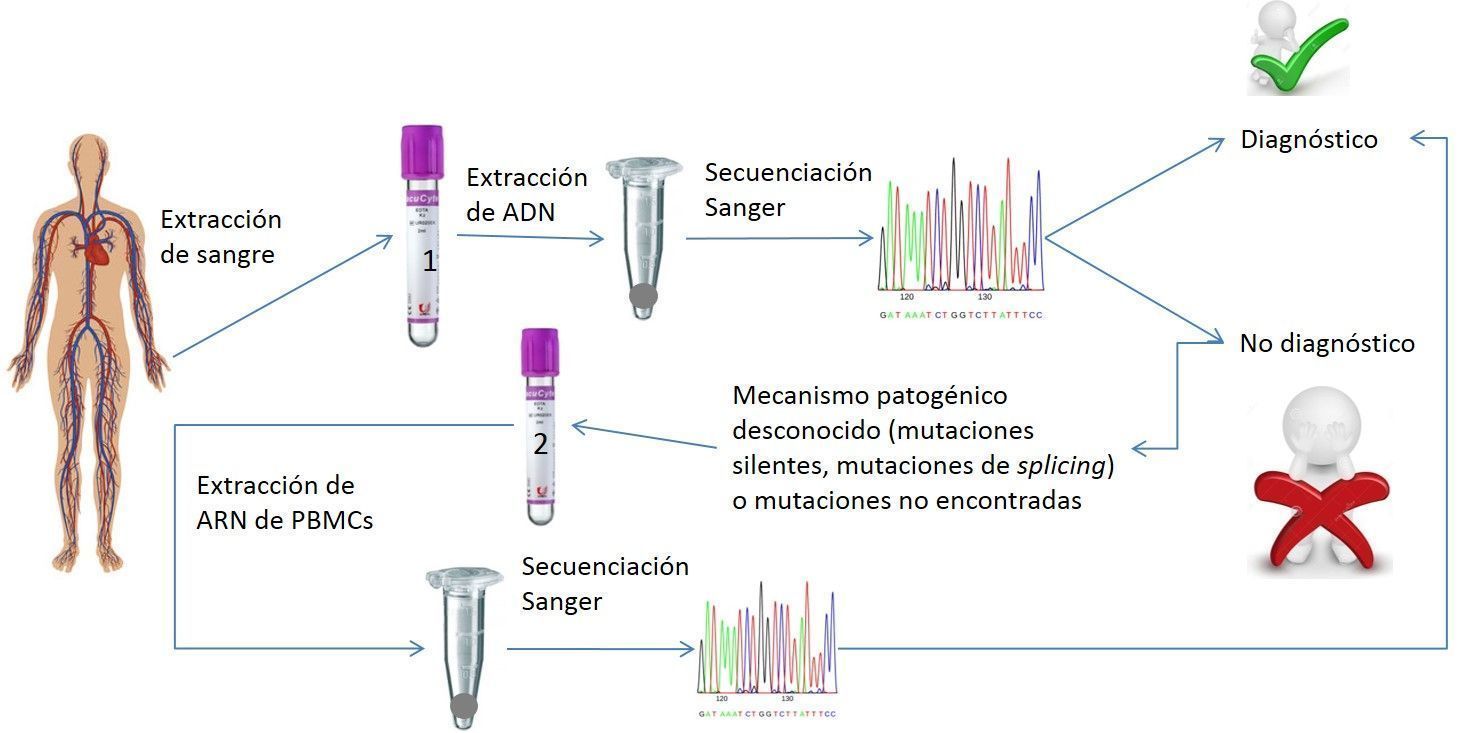
Los resultados mostraron que todas las mutaciones encontradas en el músculo fueron también detectadas en PBMCs. Además, en las PBMCs fue posible detectar mutaciones que en el músculo no habían sido identificadas, como es el caso de la mutación sin sentido p.R50*. Este resultado se explica a través del mecanismo “nonsense-mediated mRNA decay” (NMD), por el cual se eliminan transcritos aberrantes que contienen mutaciones sin sentido o que rompen la pauta de lectura, previniendo así su acumulación, y cuyo papel ya había sido descrito en el tejido muscular de los pacientes de McArdle (Nogales-Gadea et al., 2008). Este mecanismo no parece actuar en las PBMCs, posiblemente debido a la baja expresión del gen PYGM en estas células. Por este motivo, en las PBMCs pudimos caracterizar por primera vez una mutación c.645G>A, que produce una inserción total del intrón 6 en el transcrito, adicionando 91 pares de bases y alterando la pauta de lectura. Debido al NMD, este transcrito era degradado en el músculo impidiendo su detección y caracterización.
El uso de PBMCs resulta ser un método más sensible para detectar y caracterizar este tipo de mutaciones. Sabemos que la presencia de miofosforilasa es muy baja o nula en estas células, ya que no se detectaron niveles de la enzima en PBMCs de controles sanos, aun utilizando grandes cantidades de proteína total en western blot. Aun así, y a pesar de que no conocemos qué papel tiene esta transcripción residual de la miofosforilasa en PBMCs, resulta muy útil para una caracterización adecuada de algunos transcritos específicos del músculo.
A partir de los resultados de nuestro trabajo recomendamos para el diagnóstico de estos pacientes, que los laboratorios incorporen el estudio del ARN de PBMCs, en aquellos pacientes cuyas mutaciones no se encuentren a nivel de ADN, o cuyos cambios genéticos encontrados sean de significado incierto. Proponemos la obtención de dos tubos de sangre EDTA, uno para el análisis genómico directo y otro para el estudio de los ARNs en PBMCs. El método propuesto será útil para diagnosticar más casos, y contribuirá a incrementar el conocimiento de la patogenicidad de las mutaciones en la enfermedad de McArdle.
Referencia:
Garcia-Consuegra I, et al. Taking advantage of an old concept, «illegitimate transcription», for a proposed novel method of genetic diagnosis of McArdle disease. Genet Med. 2016 Feb 25. doi: http://dx.doi.org/10.1038/gim.2015.219
Bibliografía:
Nogales-Gadea G, et al. Genes and exercise intolerance: insights from McArdle disease. Physiol Genomics 2016;48:93–100. Doi: 10.1152/physiolgenomics.00076.2015
Lucia A, et al. Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. J Neurol Neurosurg Psychiatry 2012; 83:322–328. Doi: 10.1136/jnnp-2011-301593.
Nogales-Gadea G, et al. McArdle disease: update of reported mutations and polymorphisms in the PYGM gene. Hum Mutat 2015;36:669–678. Doi: 10.1002/humu.22806
García-Consuegra I, et al. Novel mutations in patients with McArdle disease by analysis of skeletal muscle mRNA. J Med Genet 2009;46:198–202. Doi: 10.1136/jmg.2008.059469
Nogales-Gadea G, et al. Expression of the muscle glycogen phosphorylase gene in patients with McArdle disease: the role of nonsense-mediated mRNA decay. Hum Mutat 2008;29:277-283. Doi: 10.1002/humu.20649

