Blanca Diaz-Castro, UK Dementia Research Institute at The University of Edinburgh.
[tabgroup title=»»]
[tab title=»Español»]
La enfermedad de Huntington es una enfermedad neurodegenerativa hereditaria y letal producida por una mutación en el gen denominado huntingtina (HTT). Mutaciones en HTT afectan gravemente al cerebro, llevando a muerte neuronal severa en el estriado y en otras zonas del cerebro, como la corteza, en menor medida. Los pacientes con enfermedad de Huntington presentan síntomas motores, psiquiátricos y cognitivos que avanzan progresivamente hasta la muerte, la cual ocurre aproximadamente 20 años después del diagnóstico. No existe tratamiento disponible para estos pacientes.
A pesar de que la causa genética de la enfermedad de Huntington se conoce, las consecuencias biológicas de esta mutación todavía no se comprenden por completo. La huntingtina mutante se expresa en neuronas pero también en células no neuronales como los astrocitos. Los astrocitos están en íntimo contacto con las neuronas y realizan una amplia variedad de funciones que incluyen el mantenimiento de la homeóstasis neuronal, la regulación de la señalización neuronal y la respuesta a daño cerebral. Se ha observado que algunas de estas funciones se alteran en la enfermedad de Huntington. Sin embargo, numerosas cuestiones surgen a partir de los estudios previos (Khakh BS et al, 2017): ¿Cuál es sustrato molecular para el desarrollo de estos fenotipos en astrocitos? ¿Qué otras alteraciones se producen en estas células? ¿Es la alteración de los astrocitos fruto de mecanismos autónomos del astrocito o depende de los cambios en otras células?
En el laboratorio del Dr. Baljit Khakh nos propusimos arrojar un poco de luz sobre estas cuestiones mediante el análisis detallado de los cambios que se producen en la expresión génica y proteica de los astrocitos en muestras de pacientes humanos y en modelos de ratón para la enfermedad de Huntington.
En un artículo, recientemente publicado en Science Translational Medicine, realizamos secuenciación de ARN de astrocitos del estriado de dos modelos de ratón con enfermedad de Huntington a tres estadios diferentes de la enfermedad: presintomática, sintomática y estadio muy avanzado. Los resultados fueron comparados con los transcriptomas de tejido completo de pacientes con enfermedad de Huntington y proteomas de ratón de la misma región cerebral, en estadios de la enfermedad equivalentes. El análisis detallado de los datos identificó los principales cambios de expresión génica en astrocitos de enfermedad de Huntington a lo largo de la progresión de la enfermedad. Utilizamos dicha información para responder las cuestiones planteadas previamente.
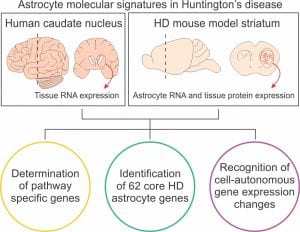
¿Cuál es el sustrato molecular para el desarrollo de fenotipos de enfermedad de Huntington en astrocitos?
Los cambios funcionales de los astrocitos en la enfermedad de Huntington descritos en estudios previos incluyen alteraciones en la señalización de calcio, la absorción de neurotransmisores, la regulación de la concentración de potasio, el metabolismo y la adquisición de un fenotipo reactivo relacionado con la neuroinflamación.
Encontramos expresión diferencial de genes implicados en el transporte de calcio, potasio y neurotransmisores que podría estar relacionada con las observaciones previas. Además, detectamos alteraciones en la expresión de genes relacionados con el metabolismo, incluyendo una disminución muy acusada en la síntesis de colesterol, así como de múltiples genes implicados en morfología y adhesión celular.
Inesperadamente, aunque se ha propuesto que la reactividad de los astrocitos podría estar causando muerte neuronal en las enfermedades neurodegenerativas, incluyendo la enfermedad de Huntington (Liddlelow SA et al, 2017), la exploración detallada de nuestros datos y los análisis de expresión en humanos no mostraron evidencias de reactividad de los astrocitos hasta los últimos estadios, más graves, de la enfermedad.
¿Qué otras alteraciones no descritas experimentan los astrocitos?
En general, el número de genes cuya expresión se altera progresa con la severidad de la enfermedad. No obstante, detectamos un solapamiento amplio de genes entre los dos modelos de ratón y los tres estadios de la enfermedad estudiados. Para algunos de estos genes, la magnitud del cambio aumentaba gradualmente con la progresión de la enfermedad. El análisis detallado de los datos reveló nuevas rutas moleculares interesantes para estudios futuros como, por ejemplo, la señalización mediante el receptor asociado a proteína G (GPCR, del inglés G protein coupled receptor), cAMP o Wnt.
Identificamos una huella de expresión génica de los astrocitos en la enfermedad de Huntington mediante la comparación de los genes de astrocitos con expresión alterada en los dos modelos de ratón con los cambios de expresión de genes en humanos y de proteínas de ratón de la misma región cerebral. Consideramos esta huella como una representación de los cambios primordiales que ocurren en los astrocitos en la enfermedad de Huntington. La huella incluye 62 genes implicados en funciones como la señalización mediada por GPCR, la señalización de calcio, la absorción de neurotransmisores y comportamientos dependientes del estriado.
¿Son las alteraciones funcionales de los astrocitos provocadas por mecanismos autónomos del astrocito o dependen de los cambios en otras células?
Para saber si las alteraciones previamente descritas se deben a los efectos de la expresión de la huntingtina en los astrocitos (mecanismos celulares autónomos) o por el contrario, son consecuencia de los cambios ocurridos en el circuito que los rodea (no autónomos), expresamos una proteína con dedos de zinc (ZFP, del inglés zinc finger protein) en astrocitos para reprimir de forma específica la expresión de la huntingtina mutante en estas células. La expresión de ZFP en astrocitos redujo los niveles de huntingtina mutante al 30% de la cantidad acumulada en los astrocitos que no expresaban ZFP. Los análisis de secuenciación de ARN de los modelos de ratón de enfermedad de Huntington que expresaban ZFP demostraron que la reducción de huntingtina mutante en astrocitos redujo la magnitud de los cambios de expresión génica de 61 de los 62 genes de la huella de expresión génica de astrocitos en la enfermedad de Huntington.
Además, encontramos que las rutas moleculares relacionadas con la enfermedad de Huntington, el daño sobre el ADN y la señalización mediada por Wnt se restauraban. Finalmente, a partir de estos experimentos identificamos a Adora2a, un GPCR capaz de regular algunos de los 62 genes de la huella molecular. La expresión de Adora2a también está alterada en los astrocitos de enfermedad de Huntington y se recupera mediante la expresión de ZFP.
En resumen, nuestros resultados definen los cambios de expresión génica en los astrocitos en enfermedad de Huntington, identifican los efectos celulares autónomos de la expresión de HTT mutante en astrocitos y desvelan rutas de señalización en estas células, como la señalización a través de GPCR, que podrían ser explotadas como dianas de tratamiento.
Bibliografía:
Khakh, B. S. et al. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017. doi:10.1016/j.tins.2017.05.002 (2017).
Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G. & Khakh, B. S. Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med. 2019. doi:10.1126/scitranslmed.aaw8546.
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature . 2017. doi:10.1038/nature21029.
[/tab]
[tab title=»English»]
Researchers define the molecular alterations of astrocytes as potential targets for treatment in the neurodegenerative disease of Huntington’s
Huntington’s disease (HD) is a hereditary and fatal neurodegenerative disease caused by a mutation in a gene called huntingtin (HTT). HTT mutations strongly affect the brain, leading to severe neuronal death in the striatum and, to a lesser extent, other brain areas like the cortex. Patients with HD present motor, psychiatric and cognitive symptoms that progressively advance until decease occurs about 20 years after diagnosis. There is no treatment available for this patients.
Despite the genetic cause of HD is known, we still don’t fully understand the biological consequences of this mutation. Mutant huntingtin is expressed in neurons, but also in non-neuronal cells like astrocytes. Astrocytes are in intimate contact with neurons and perform a wide variety of functions including neuron homeostasis maintenance, regulation of neuronal signaling and response to injury. It has been shown that some of these functions are altered in HD, however, many questions arise from previous studies (Khakh BS et al, 2017) . What is the molecular substrate for the development of such phenotypes in astrocytes? What other alterations do these cell undergo? Is astrocyte dysfunction due to cell-autonomous or non-cell-autonomous mechanisms?
In Dr. Baljit Khakh lab, we sought to shed some light on these questions by performing a detailed analysis of astrocyte gene and protein expression changes in human patients and mouse models of HD. In Diaz-Castro et al., recently published in Science Translational Medicine, we generated astrocyte specific RNA-seq datasets from striatum of two mouse models of HD at three different stages of the disease: presymptomatic, symptomatic and late stage. The results were compared to whole tissue human HD transcriptomes and mouse proteomes of the same brain region at equivalent disease stages. Deep analysis of the data identified the main gene expression changes of HD astrocytes along the progression of the disease. We used such datasets to answer the questions raised above.
What is the molecular substrate for the development of HD phenotypes in astrocytes?
Previously described functional changes of astrocytes in HD include alterations in calcium signaling, neurotransmitter uptake, potassium buffering, metabolism, morphology and acquisition of neuroinflammation related reactive phenotype. We found differential expression of genes involved in calcium, potassium and neurotransmitter transport that could underlie previous observations. We also detected alterations in metabolism related genes, including a very strong downregulation of cholesterol synthesis, as well as many genes involved in cell morphology and adhesion. Interestingly, although it has been proposed that astrocyte reactivity could be causing neuronal cell death in neurodegenerative diseases, including HD (Liddlelow SA et al, 2017), detailed exploration of our data and human expression analyses showed no evidence of astrocyte reactivity until late very severe stages of the disease.
What other unreported alterations do astrocytes undergo?
Overall, although the number of differentially expressed genes progresses with the severity of the disease, we found a high gene overlap between mouse models and disease stages. For some of these genes, the magnitude of change was gradually increased with the disease progression. Detailed analysis of the data unveiled new interesting pathways for further exploration like G-protein couple receptor (GPCR), cAMP or Wnt signaling.
By comparing the mouse astrocyte genes which differential expression was conserved, across models and ages, to human gene expression changes and mouse proteomics data of the same brain region, we identified a gene expression signature for astrocytes in HD. We consider this signature to represent the core astrocyte changes in HD. It includes 62 genes that are involved in functions like GPCR signaling, calcium signaling, neurotransmitter uptake and striatal dependent behaviors.
Are astrocyte dysfunctions due to cell-autonomous or non-cell-autonomous mechanisms?
To know if the previously described alterations were due to the effects of the expression of huntingtin in the astrocytes (cell autonomous mechanism) or instead they were a consequence of the changes occurring in the surrounding circuit (non-cell autonomous), we expressed a zinc finger protein (ZFP) in astrocytes to specifically repress the expression of mutant huntingtin in these cells. ZFP expression in astrocytes reduced the levels of mutant HTT down to 30 % of the amount in non-ZFP expressing astrocytes. RNA-seq analyses of HD mouse models that expressed ZFP demonstrated that reduction of mutant huntingtin in astrocytes successfully ameliorated the gene expression changes of 61 of the 62 HD astrocyte signature genes. In addition, we found that pathways related to HD, DNA damage and Wnt signaling were further restored. Finally, through these experiments, we identified Adora2a, a GPCR that functions as upstream regulator of the 62 signature genes which expression is altered in HD astrocytes but rescued by the ZFP expression.
In conclusion, our results define the astrocyte gene expression changes in HD, identify the cell autonomous effects of expression of mutant HTT in astrocytes and unveil potential pathways of these cells, like GPCR signaling, that could be further exploited as targets for treatment.
Bibliography
Khakh, B. S. et al. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422-437, doi:10.1016/j.tins.2017.05.002 (2017).
Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G. & Khakh, B. S. Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med 11, doi:10.1126/scitranslmed.aaw8546 (2019).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481-487, doi:10.1038/nature21029 (2017).
[/tab]
[/tabgroup]
Si te ha gustado esta noticia y quieres aprender más sobre Genética y Genómica aplicadas a la salud, te interesan nuestros cursos. Tienes más información aquí.



