Maria Aguilar Ballester
Investigadores del Baylor College of Medicine (Houston), han realizado exitosamente el primer tratamiento efectivo de una enfermedad genética autosómica dominante cardiaca por edición genética somática mediante el sistema CRISPR-Cas9 en una línea de ratón portadora de una mutación concreta hallada en un paciente.

En la última década hemos sido testigos de los grandes avances realizados en la aplicación del sistema CRISPR-Cas9 como herramienta de modificación genética en cultivos celulares y modelos animales. Si bien hace poco saltaba la noticia de los primeros seres humanos modificados genéticamente durante su gestación, y con ello el correspondiente debate ético, lo cierto es que hace años que se investiga la empleabilidad del sistema de modificación genética basada en CRISPR como posible tratamiento para organismos completamente desarrollados aquejados de alguna enfermedad genética. Es el caso de los grupos de investigación del Dr. Xander Wehrens y de Dr. William Lagor, del Baylor College of Medicine (Houston), quienes han reenfocado el uso de esta técnica como un posible tratamiento aplicable en medicina personalizada para la cura de enfermedades genéticas actualmente incurables.
Cada año mueren aproximadamente 3 millones de personas en el mundo debido a enfermedades cardiacas, muchas de ellas de origen genético hereditario. Por ello los investigadores han tomado el caso de un paciente afectado de taquicardia ventricular polimórfica catecolaminérgica (CPVT) hereditaria, en cuya familia existían antecedentes de muerte súbita debido a esta misma patología. En esta enfermedad la tasa de mortalidad es del 50% en los 8 años tras el diagnóstico y ningún tratamiento de los existentes es completamente efectivo, además de presentar frecuentes efectos secundarios.
La causa genética más frecuente de la CPVT, como en el presente caso, se debe a mutaciones autosómicas dominantes en el gen codificante del receptor de rianodina 2 (RYR2). El canal formado por RyR2 es el responsable de la liberación de Ca2+ del retículo sarcoplásmico que inicia la contracción del miocito, en respuesta a la entrada masiva de Ca2+ al citoplasma a través de canales dependientes de voltaje. Con solo una subunidad mutada en el homotetrámero que forma, ya es suficiente para que se produzcan liberaciones espontáneas de Ca2+ al citoplasma, incluso tras concluir la despolarización, que ocasionan graves arritmias de resultado posiblemente mortal.
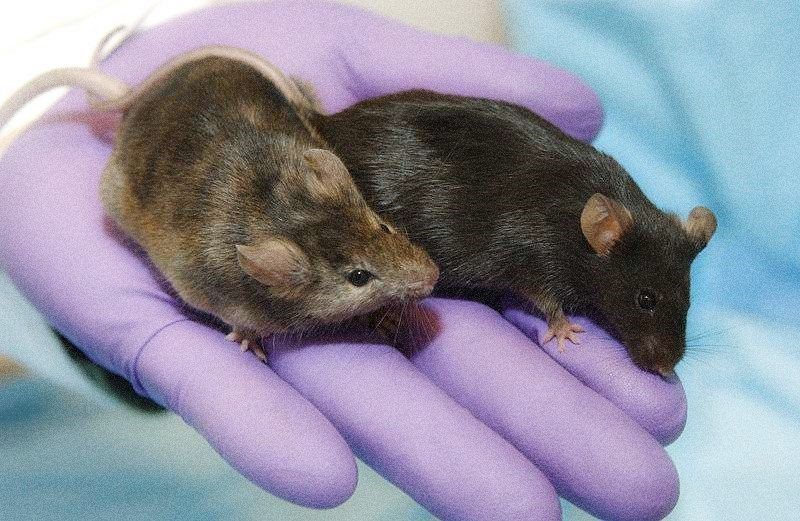
El paciente de este estudio era portador en heterocigosis de una mutación de cambio de aminoácido (Los investigadores quisieron probar la aplicabilidad terapéutica de la técnica de edición genética para introducir una mutación en el alelo mutado que truncara la proteína mutante. Para ello, crearon una línea de ratón portadora de la misma mutación en heterocigosis (R176Q/+) y 10 días tras el nacimiento les inyectaron subcutáneamente el sistema CRISPR-Cas9 mediante un vector AVV dirigido específicamente por el ARNg (RNA guía cuya secuencia aparea específicamente con la secuencia diana) al alelo mutado.
Los investigadores observaron efectividad y seguridad de la terapia génica al restablecer la actividad cardiaca normal por reducción de los niveles de ARNm y proteína de la subunidad mutada del canal sin afectar a los flujos de Ca2+ más allá del causante de la enfermedad y sin mostrar ediciones genéticas fuera de la secuencia diana del alelo mutado.
Sin embargo, el estudio todavía muestra limitaciones. Una de ellas es que el RNA guía todavía no se une directamente a la secuencia mutada sino a una secuencia adyacente artificial. Otra limitación es la idoneidad de nuevos modelos preclínicos para poder testar la reproducibilidad del tratamiento antes de poder testarlo en humanos. Recientemente, el Dr. Xander Wehrens declaró: “ahora estamos testando la misma aproximación en células madres de pacientes con la misma condición, con el fin de analizar la eficacia y seguridad en células humanas”.
No obstante, no por ello deja de constituir un interesante y prometedor avance, no solo en el ámbito de las enfermedades genéticas cardiacas, sino también del futuro de los tratamientos basados en edición genética y la medicina personalizada. En palabras del Dr. Xander Wehrens: “creemos que la edición de precisión del genoma es el futuro de las terapias génicas dirigidas a tejidos, y las enfermedades cardiacas severas son un buen punto desde el que empezar”.
Referencia: Pan X, et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation Res. 2018. Doi: http://dx.doi.org/10.1161/CIRCRESAHA.118.313369
Fuente: Preventing sudden cardiac death with genome editing. https://blogs.bcm.edu/2018/10/30/preventing-sudden-cardiac-death-with-genome-editing/
Si te ha gustado esta noticia y quieres aprender más sobre Terapia Génica, quizás te interese nuestro curso “Introducción a la Terapia Génica y Edición del Genoma”. Tienes más información sobre el curso aquí.