
Amparo Tolosa, Genética Médica News
Un estudio dirigido por el Massachusetts General Hospital ha conseguido recuperar la expresión génica del cromosoma X inactivado para tratar, en ratón, el Síndrome de Rett.
Hombres y mujeres nacen con 22 pares de cromosomas autosómicos, comunes para ambos, y una pareja de cromosomas sexuales diferente para cada sexo: las mujeres nacen con dos cromosomas X en cada célula y los hombres con un cromosoma X y un cromosoma Y. Como las células sólo necesitan un cromosoma X activo, durante el desarrollo existe un mecanismo que inactiva de forma aleatoria y permanente uno de los cromosomas X en cada célula femenina. Una vez inactivado un cromosoma X, las células hijas heredan el mismo patrón de inactivación en las siguientes divisiones.
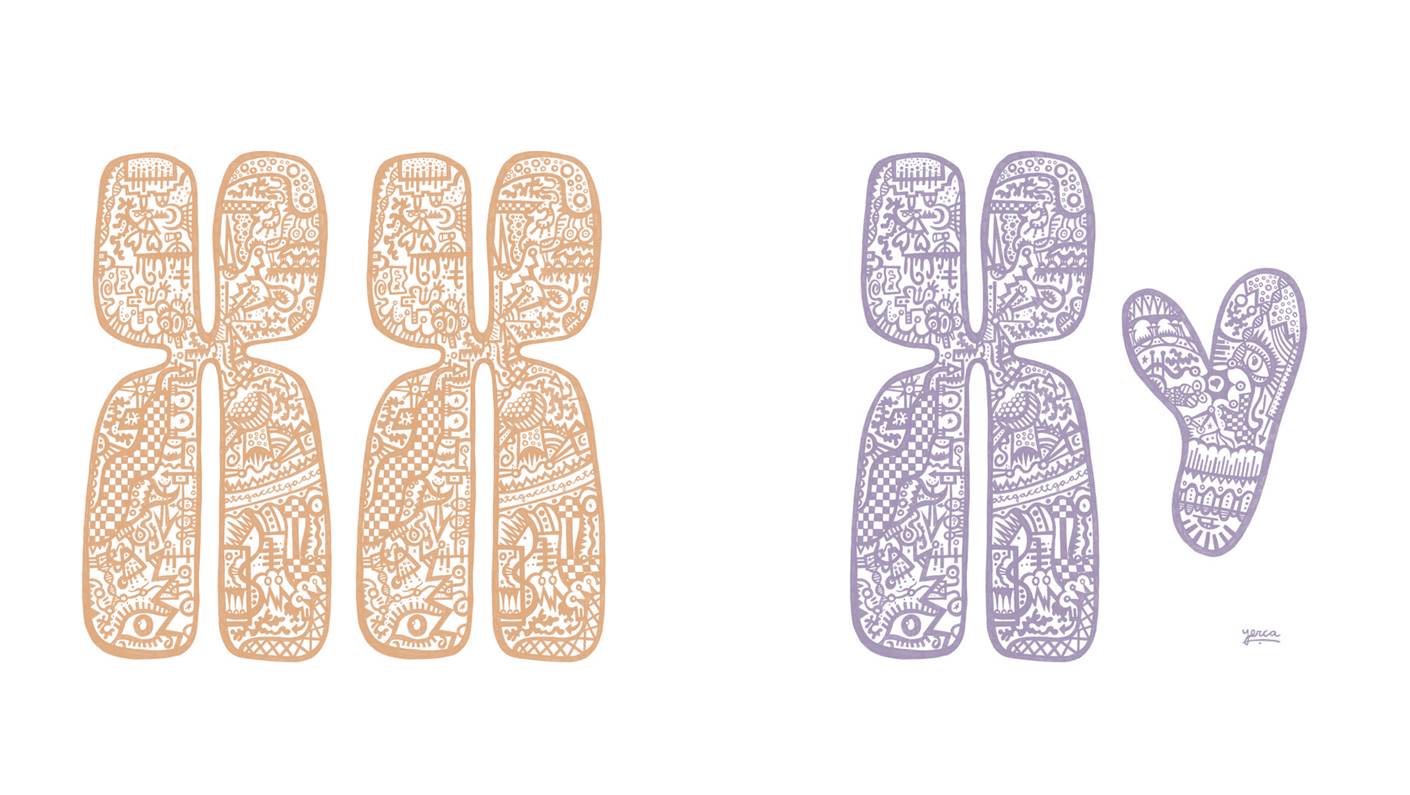
La inactivación del cromosoma X de forma aleatoria en diferentes células femeninas lleva a que las mujeres tengan células en las que un cromosoma X está inactivado y células en las que el inactivado es el otro. Si uno de los cromosomas X tiene una mutación que afecta a su función, las células en las que la copia normal se expresa pueden compensar a veces la pérdida de función de aquellas en las que el cromosoma activo es el que contiene la mutación. Como resultado, el efecto de muchas enfermedades causadas por mutaciones en genes del cromosoma X es mayor en varones, que manifiestan el efecto de la única copia de cromosoma X que tienen. Es el caso, por ejemplo de la hemofilia. En otros casos, como el Síndrome de Rett o el síndrome del X frágil, no ocurre así y la presencia de células con copias normales del cromosoma X no es suficiente para compensar la ausencia de función en las células en las que la copia con la mutación está activa.
Una paradoja del Síndrome de Rett es que las pacientes llevan la cura (el cromosoma X que lleva la copia normal del gen) en sus propias células, pero ésta está inactivada. Así, un potencial tratamiento para estas enfermedades ligadas al cromosoma X sería la reactivación del gen de la copia sana. Con este objetivo un equipo de investigadores del Massachusetts General Hospital ha desarrollado un sistema para activar el cromosoma X inactivado y restaurar la expresión de genes como el que causa el Síndrome de Rett. La prueba de concepto ha sido realizada en células de ratón. Sin embargo, los resultados sugieren que la reactivación podría obtenerse también en el cerebro y ser utilizada para el tratamiento de diferentes enfermedades asociadas al cromosoma X.
“La aproximación descrita en nuestro trabajo aprovecha el hecho de que cada paciente lleva la cura dentro de sus propias células, pero esa cura está bloqueada por un proceso que dura la vida entera denominado inactivación del cromosoma X,” señala Jeannie T Lee, profesora de Genética en la Universidad de Harvard y directora del trabajo. “Nuestro objetivo ha sido desbloquear el cromosoma X inactivo y restaurar la expresión de la copia buena del gen.”
El síndrome de Rett es un trastorno cerebral que se presenta de forma casi exclusiva en niñas. Las pacientes muestran un desarrollo normal inicial. Sin embargo, a partir de los 12 o 15 meses comienzan a manifestar problemas en el lenguaje, aprendizaje y otras características neurológicas. La mayor parte de los casos de Síndrome de Rett están causados por la presencia de mutaciones en el gen MECP2, localizado en el cromosoma X. MECP2 codifica para una proteína reguladora de la expresión génica que participa en diversas funciones de las células nerviosas y cuando su función está alterada en uno de los cromosomas X se compromete la comunicación entre ellas.
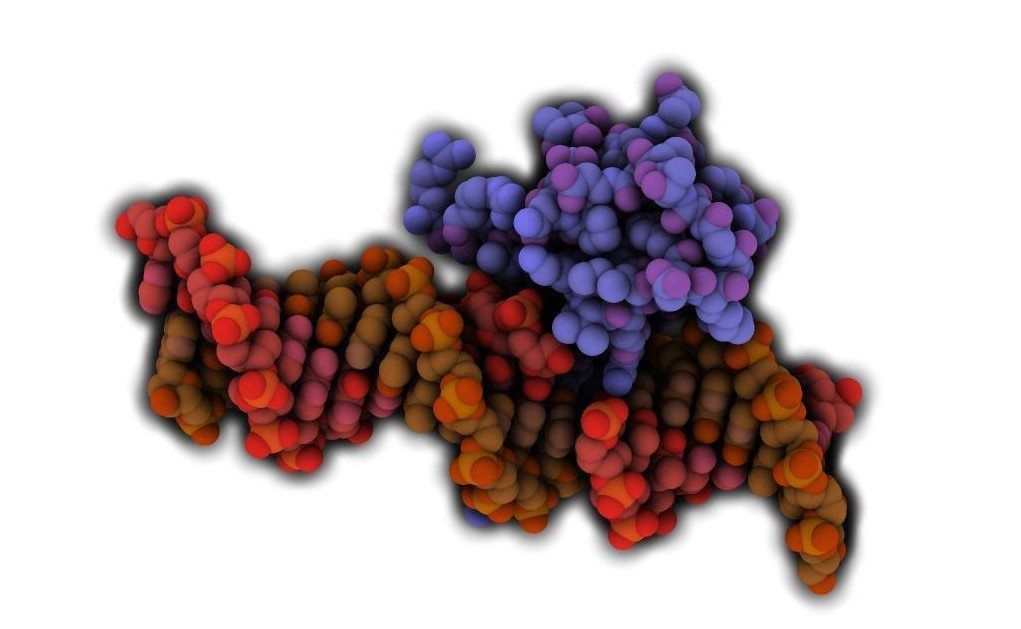
La proteína MECP2 interaccionando con el ADN. Imagen: Protein Data Base- 3C2I, visualizada con QuteMol (http://qutemol.sourceforge.net).
La inactivación del cromosoma X está mediada por un ARN no codificante expresado en el propio cromosoma inactivado, conocido como Xist. Este ARN es esencial para que se inicie el proceso de inactivación y para que su acción se propague a lo largo de todo el cromosoma. Los investigadores bloquearon la acción de Xist en células de ratón mediante la utilización de oligonucleótidos antisentido dirigidos de forma específica hacia Xist. Los oligonucleótidos reconocen la molécula Xist, se unen a ella y facilitan su degradación. Además, en paralelo, los investigadores utilizaron un inhibidor de la metilación del ADN, denominado Aza (5-aza-2′-deoxycytidina). La suma de ambos tratamientos permitió aumentar la expresión de MECP2 hasta 30.000 veces en dos líneas celulares diferentes de ratón.
Para comprobar que la estrategia combinada no resultaría tóxica en un sistema in vivo los investigadores suprimieron la expresión de Xist en cerebro. Los ratones obtenidos, y concretamente las hembras, no mostraron efectos negativos visibles. Además, puesto que el tratamiento a largo plazo con Aza resulta tóxico, los investigadores lo utilizaron para tratar los ratones de forma puntual. En estos ratones, la supresión de Xist se tradujo en cierta reactivación del cromosoma X y únicamente al tratar los animales con Aza se consiguió reactivar el cromosoma X inactivo de forma específica.
“Los fármacos de oligonucleótidos antisentido son muy selectivos, puesto que deben coincidir exactamente con la molécula de ARN frente a la que están dirigidos, lo que reduce la probabilidad de que ocurran efectos negativos, y puesto que Xist tiene un papel muy específico en la inactivación del cromosoma X ningún otro proceso celular debería ser alterado,” manifiesta Lieselot Carrette, primer autor del trabajo e investigador en el Departamento de Biología del Massachusetts General Hospital. “El inhibidor de la metilación Aza es menos selectivo pero la sinergia producida en combinación con el fármaco antisentido de Xist nos permite utilizar relativamente bajas dosis, lo que hace que su combinación sea una opción asequible.”
Los resultados del trabajo proporcionan una prueba de concepto para la reactivación del cromosoma X inactivo como aproximación para el tratamiento de enfermedades causadas por mutaciones en el cromosoma X como el síndrome de Rett. Los investigadores confían en poder obtener mejores modelos de la enfermedad para poder comprobar in vivo la eficacia del tratamiento combinado de los oligonucleótidos antisentido y el inhibidor de la metilación Aza.
“Esta es un área activa de investigación y nuestros datos preliminares indican que no es necesaria la expresión completa de la proteína MECP2 para obtener una mejora de los síntomas,” manifiesta Lee. “Por tanto, somos optimistas de que nuestra aproximación proporcionará eventualmente un tratamiento significativo para los pacientes, aunque queda mucho trabajo por hacer.”
Investigación original: Carrette LLG, et al. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc Natl Acad Sci U S A. 2017 Dec 27. doi: http://dx.doi.org/10.1073/pnas.1715124115
Fuente: X chromosome reactivation could treat Rett syndrome, other X-linked disorders. http://www.massgeneral.org/about/pressrelease.aspx?id=2195

