Marta Alonso-Peña1, María J. Monte1,3, Oscar Briz1,3, Elisa Herráez1,3, Carmen Berasain2, 3, Josep M. Argemi2, 3, Jesús Prieto2, 3, José J.G. Marín1, 3
1)Laboratorio de Hepatología Experimental y Vectorización de Fármacos (HEVEFARM), Instituto de Investigación Biomédica de Salamanca (IBSAL), Universidad de Salamanca.
2)Departamento de Medicina, Clínica Universidad de Navarra, Centro de Investigación Médica Aplicada (CIMA), Universidad de Navarra.
3)Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd).
En un estudio desarrollado por investigadores pertenecientes al Centro de Investigación Biomédica en Red para el Estudio de Enfermedades Hepáticas y Digestivas (CIBERehd) englobados en dos equipos, uno de la Universidad de Salamanca y el Instituto de Investigación Biomédica de Salamanca (IBSAL), y otro del Centro de Investigación Médica Aplicada de la Universidad de Navarra (CIMA), se ha identificado una nueva entidad nosológica (OMIM #617308), debida a un defecto congénito parcial en la enzima peroxisomal ACOX2, implicada en el metabolismo de los ácidos biliares. Esta nueva patología denominada “deficiencia de ACOX2” dificulta la síntesis normal de ácidos biliares y determina la acumulación de especies moleculares intermedias con potencial hepatotoxicidad (Monte, 2017).
La elevación de los niveles de transaminasas en suero es un marcador muy utilizado en bioquímica clínica que indica la existencia de daño hepático, el cual puede ser causado por una gran variedad de patologías. Sin embargo, en alrededor de un 15% de los pacientes en los que se detecta hipertransaminasemia el origen permanece sin identificar (Cacciola, 2017). Establecer la etiología de esta alteración es crucial para su correcto tratamiento, ya que la agresión mantenida del tejido hepático puede predisponer a largo plazo al desarrollo de enfermedades tan serias como la cirrosis e incluso el cáncer hepático.
El estudio partió del caso clínico de un paciente adolescente aparentemente sano y deportista que, tras una lesión articular, sufrió una crisis de hepatotoxicidad asociada al tratamiento con analgésicos y antiinflamatorios no esteroideos. Tras este episodio, y sin causa aparente, los niveles séricos de transaminasas se mantuvieron elevados de forma persistente durante 2 años. El tratamiento del paciente con colestiramina, una resina administrada por vía oral que a su paso por el intestino secuestra los ácidos biliares favoreciendo así su eliminación, consiguió normalizar los niveles de transaminasas, lo que hizo sospechar que pudiera tratarse de una alteración en el metabolismo de los ácidos biliares.
El análisis por espectrometría de masas del suero y la orina del paciente reveló niveles extremadamente bajos de ácidos biliares maduros, así como la presencia de un metabolito intermedio de la síntesis de estos compuestos, el ácido tauro-trihidroxicolestanoico (tauro-THCA). Por otra parte, a partir del ADN del paciente se amplificaron por PCR de alta fidelidad y se secuenciaron todos los exones de las enzimas que podrían ser responsables de la acumulación de este metabolito. Tan solo se encontró una alteración sospechosa de causar un cambio de función: una mutación en homocigosis (c.673C>T) que ocasiona un cambio de aminoácido (p.R225W) en la enzima peroxisomal ACOX2, implicada en el acortamiento de la cadena lateral del THCA.
El estudio genético de los familiares del paciente reveló que tanto el padre como la madre eran portadores heterocigotos de la mutación, aunque no presentaban ningún indicio de alteración hepática. En cambio, la hermana, dos años menor que el paciente, también era portadora de la mutación c.673C>T en homocigosis. Su análisis bioquímico mostró niveles séricos muy bajos de especies moleculares maduras de ácidos biliares junto con una acumulación de tauro-THCA, así como una historia clínica con picos de hipertransaminasemia asociados a la utilización de analgésicos y antiinflamatorios.
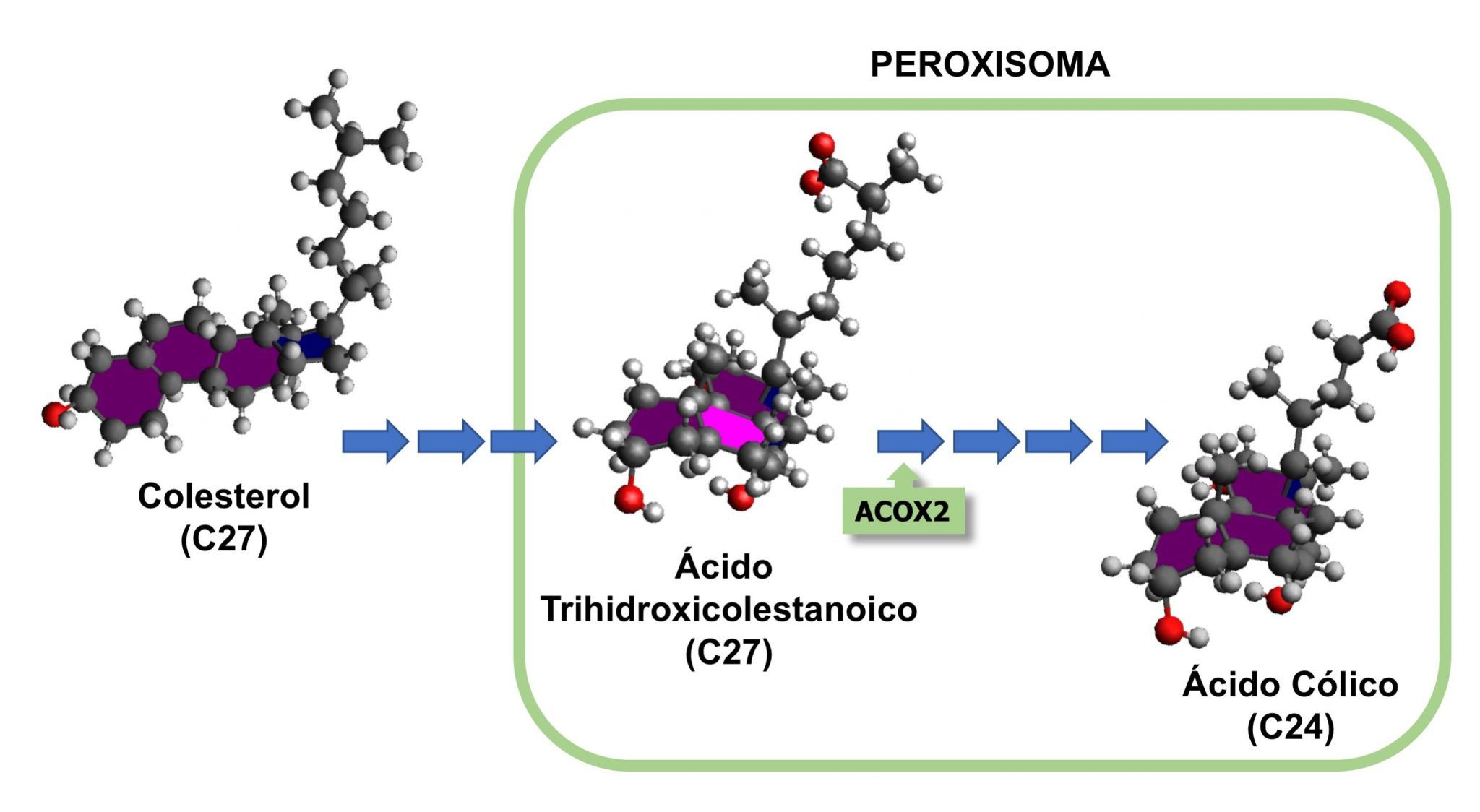
Con objeto de investigar la repercusión funcional de la mutación encontrada, se desarrolló un modelo in vitro de la enfermedad. Para ello, se obtuvieron sublíneas celulares de hepatoblastoma humano que expresaban establemente la variante silvestre del gen (ACOX2) o la mutada (ACOX2-R225W). Mediante western blot e inmunofluorescencia se observó que la mutación no alteraba ni el tamaño de la proteína ni su localización peroxisomal. La síntesis de ácidos biliares se investigó incubando estas células con THCA. En células que expresaban ACOX2-R225W, la formación de ácido cólico a partir de THCA se reducía en un 94% respecto a las que expresaban ACOX2, detectándose también la acumulación del precursor. Además, en las células que expresaban ACOX2-R225W, la exposición al THCA causó un marcado estrés oxidativo y una reducción de la viabilidad celular. Estos efectos tóxicos se prevenían si en las células se sobre-expresaba la variante silvestre de ACOX2.
Así pues, se ha identificado un nuevo defecto genético en la vía de síntesis de ácidos biliares, que afecta a la enzima peroxisomal ACOX2. Este tipo de patologías se incluyen en el espectro del síndrome de Zellweger (Waterham, 2016), para las cuales se ha demostrado que la terapia de reemplazamiento con ácidos cólico, quenodesoxicólico y/o ursodesoxicólico (Berendse, 2016) es beneficiosa para activar los mecanismos hepáticos que limitan la síntesis de novo de ácidos biliares, y en este caso del metabolito tóxico THCA, evitando así la continua agresión al parénquima hepático. Por tanto, el descifrar las bases moleculares de esta afección ha proporcionado información esencial para la detección precoz del trastorno en pacientes con hipertransaminasemia asintomática, así como para la elección del tratamiento más adecuado que permita prevenir lesiones hepáticas más serias en individuos portadores de esta mutación en homocigosis.
Referencia: Monte MJ, et al. ACOX2 deficiency: An inborn error of bile acid synthesis identified in an adolescent with persistent hypertransaminasemia. J Hepatol. 2017 Mar;66(3):581-588. doi: http://dx.doi.org/10.1016/j.jhep.2016.11.005
Bibliografía:
Cacciola I, et al. Evaluation of liver enzyme levels and identification of asymptomatic liver disease patients in primary care. Intern Emerg Med (2017) 12:181–186. doi:10.1007/s11739-016-1535-2
Berendse K, et al. Cholic acid therapy in Zellweger spectrum disorders. J Inherit Metab Dis (2016) 39(6):859-868. doi: 10.1007/s10545-016-9962-9
Online Mendelian Inheritance in Man. URL: https://omim.org/entry/617308 [22/03/2017]
Waterham HR, et al. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta (2016) 1863(5):922-33. doi: 10.1016/j.bbamcr.2015.11.015

