
Maria Aguilar Ballester
El grupo de investigación de Ethan Greenblatt y Allan Spradling del Departamento de Embriología de la Carnegie Institution for Science ha publicado recientemente en la revista Science sus últimos hallazgos acerca de las bases genéticas subyacentes al fallo ovárico prematuro y el síndrome del X frágil, el tipo más frecuente de desorden dentro del rango autista. Empleando como modelo ovocitos maduros de Drosophila han sido capaces de relacionar ambos desórdenes con la pérdida de función del gen fragile X mental retardation gene (FMR1), sin el cual se observan graves defectos para traducir ARNm codificantes de proteínas de extraordinario gran tamaño.
El síndrome del X frágil, el tipo más común de discapacidad intelectual y trastorno dentro del espectro autista, está causado por mutaciones en el fragile X mental retardation gene (FMR1). La proteína codificada por el gen forma complejos ribonucleoproteicos (RNP) e incorpora ARNm almacenados en el interior celular a sistemas poliribosómicos para su traducción. Dicha función resulta crucial en el caso de los tejidos neuronal y ovárico, donde en condiciones normales se controla a nivel traduccional la expresión de muchos genes, algunos de ellos claves en los correctos desarrollo y funcionamiento de los tejidos, como se ha comprobado en humanos, ratón y Drosophila.
No obstante, todavía no se han identificado dichos transcritos diana de FMR1. El principal problema hasta ahora había sido la dificultad de establecimiento de un modelo de estudio, así como la extracción, a partir de tejido neuronal, de muestras de RNPs que contuvieran FMR1 en una concentración aceptable para su estudio.
Ethan Greenblatt y Allan Spradling han hallado en los ovocitos maduros de Drosophila un estado fisiológico idóneo como modelo de estudio. En ellos la carencia de transcripción otorga al proceso de traducción el control de los procesos que inicie el ovocito durante su almacenamiento y posterior inicio del desarrollo embrionario, tal y como sucede en tejido nervioso y ovárico.
Para los experimentos, los ovocitos maduros fueron inducidos a un estado de quiescencia dentro de sus respectivos folículos, tras el cual se procedía a la inducción de su ovulación y fecundación. Los embriones así obtenidos eran comparados en función de la actividad de FMR1 y la duración del estado de quiescencia al que habían sido sometidos antes a la fecundación.
En primer lugar, los investigadores observaron que la viabilidad de los embriones fecundados tras 10 días en estado de quiescencia del ovocito era de un 80% en el caso de la mayoría de líneas, incluida la línea control, pero descendía a entre un 20-30% en la línea donde la traducción de FMR1 era inhibida por ARNi, diferencia que no era apreciable en ovocitos almacenados un día o que maduraban y se fecundaban en la misma madre tras el experimento. En palabras de Allan Spradling: “una reminiscencia de la insuficiencia ovárica humana”.
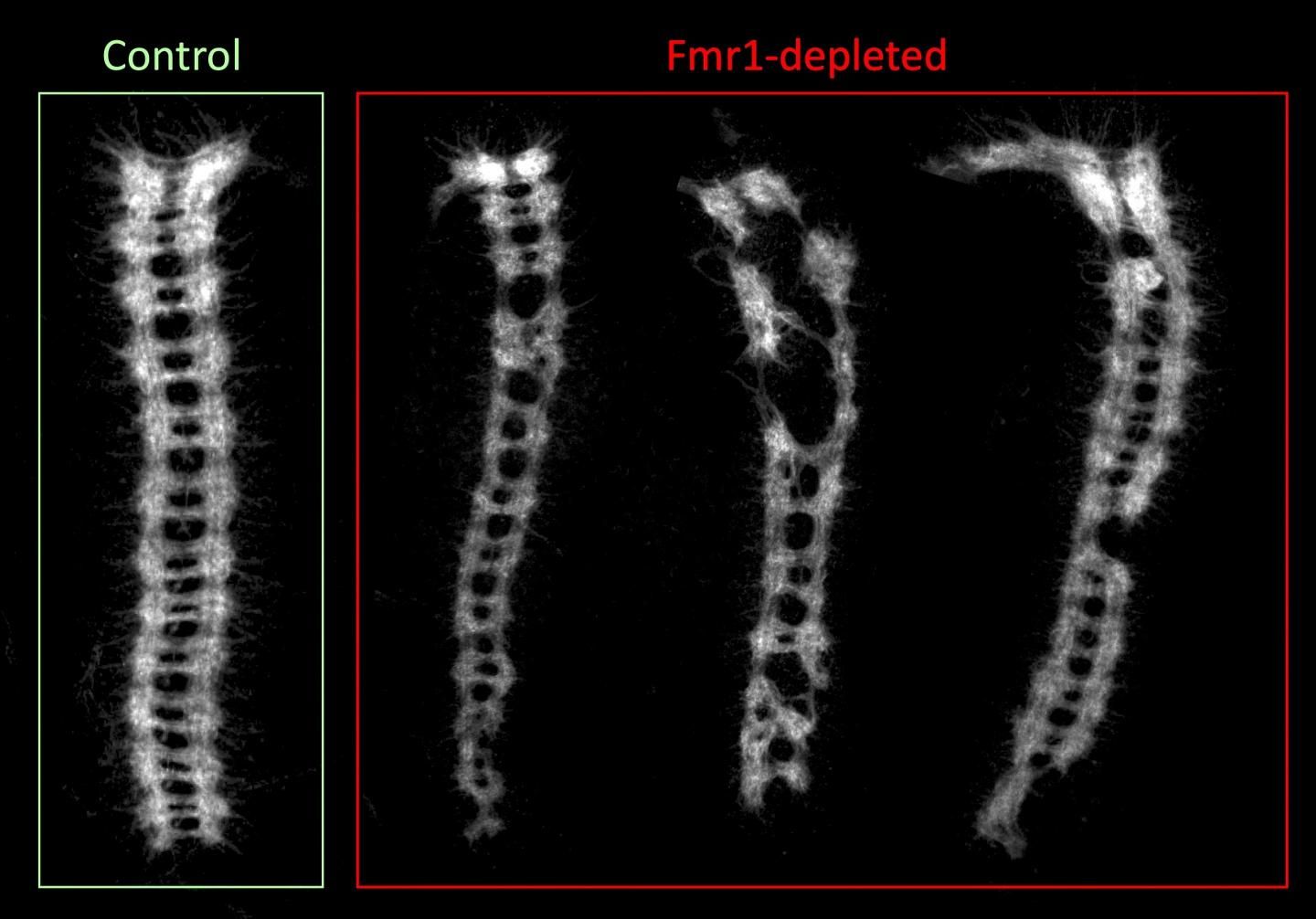
Paralelamente, se realizó un marcaje específico del cordón nervioso ventral sobre los embriones con el fin de estudiar su efecto en el correcto desarrollo del sistema nervioso. Así se comprobó que en este caso también se desarrollaban diferentes al resto los embriones desarrollados a partir de ovocitos fecundados tras 10 días en quiescencia pertenecientes a la línea donde FMR1 se encontraba reprimido. Más del 50% de los embriones no eran capaces de desarrollar correctamente el sistema nervioso, sin que este hecho guardara relación con el ya mencionado descenso en la viabilidad del embrión, pues embriones formados a partir de ovocitos de la línea control almacenados hasta 14 días veían reducida su viabilidad a un 20% pero seguían sin mostrar alteraciones en el desarrollo del sistema nervioso del embrión.
A continuación se realizó un estudio del perfil ribosómico con el fin de evaluar la traducción del genoma completo. Los resultados revelaron niveles normales de ARNm. Por tanto, la represión de FMR1 no afecta a la transcripción ni a la estabilidad de los transcritos. Sin embargo, 421 genes se encontraban subtraducidos, 56 considerados homólogos de genes humanos implicados en síndromes humanos del neurodesarrollo, principalmente tumores neuronales y desórdenes cognitivos y de conducta.
Para finalizar y con el fin de resolver más detalles acerca del funcionamiento de la proteína FMR1, se realizó sobre muestras de tejido cerebral de Drosophila melanogaster un protocolo de formación de enlaces entre FMR1 y los ARNm con los que está interactuando, seguido de una inmunoprecipitación y posterior análisis de las secuencias inmunoprecipitadas unidas a FMR1. Los resultados permitieron concluir que la proteína FMR1 establece contacto directo y potencia la traducción, especialmente de transcritos con tamaño proteico o longitud UTR muy superiores a la media en Drosophila. Estos mismos transcritos podrían codificar las proteínas responsables de la inhibición traduccional o catálisis de aquellos transcritos que muestran un aumento en su traducción cuando FMR1 es reprimido. Los genes reprimidos por FMR1 suelen mostrar una correlación negativa con la estabilidad proteica, la traducción o el crecimiento celular. Entre las secuencias diana confirmadas destaca el gen Poe/Ubr4, que codifica de una proteína de gran tamaño cuya función se demostró firmemente ligada al desarrollo de sistema nervioso pero no a la viabilidad del embrión.
Este estudio supone una mejora notable en la compresión de trastornos asociados a diversos tejidos dependientes del almacenamiento y regulación precisa de la traducción de ARNm, destacando aquellos que afectan a tejidos neuronales, ovocitos y espermatocitos, trastornos cada vez más frecuentes en la sociedad actual por el efecto que tiene en ellos el aumento de la esperanza de vida.
La propuesta de Drosophila como organismo modelo de efectividad probada para el estudio de la pérdida de función del gen FMR1 permitirá ahondar en sus mecanismos funcionales e identificar dianas de su función, a partir de las cuales se podrían identificar posibles dianas terapéuticas y desarrollar múltiples estrategias de tratamiento para un amplio rango de enfermedades. En palabras de Ethan Greenblatt: “lo que más interesante me resulta es la posibilidad de estudiar el desarrollo del ovocito en el contexto de terapias potenciales que puedan revertir el efecto de perder [la función de] FMR1 desde un interés clínico”.
Referencia: Greenblatt EJ, Sprading AC, et al. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science. 2018. Doi: http://dx.doi.org/10.1126/science.aas9963
Fuente: Autism linked to egg cells’ difficulty creating large proteins. https://carnegiescience.edu/news/autism-linked-egg-cells%E2%80%99-difficulty-creating-large-proteins



