
Laura Remacha1, Mercedes Robledo1,2 y Alberto Cascón1,2
1Grupo de Cáncer Endocrino Hereditario, Programa de Genética del Cáncer Humano, Centro Nacional de Investigaciones Oncológicas (CNIO), Madrid, España.
2Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, España.
Los feocromocitomas (PCC, del inglés pheochromocytoma) y los paragangliomas (PGL), son tumores neuroendocrinos poco frecuentes considerados como una única enfermedad (PPGL para referirse a ambos tumores), pero que difieren en características clínicas tales como la localización anatómica, la secreción de catecolaminas y el potencial metastásico. Los PCCs son tumores adrenales, generalmente productores de catecolaminas y benignos, mientras que los PGLs pueden ser o no funcionales o metastásicos dependiendo de que se localicen en los paraganglios de la región toraco-abdominal o en los de la cabeza y el cuello, respectivamente.
Se estima que en torno a un 40% de los pacientes con PPGL presentan mutaciones germinales en alguno de los más de 15 genes de susceptibilidad conocidos, siendo por tanto el tumor con el mayor componente hereditario conocido hasta la fecha (Dahia et al, 2014). Sin embargo, todavía queda un porcentaje considerable de pacientes que, si bien no portan mutaciones en ninguno de los genes conocidos, muestran características clínicas propias de una enfermedad hereditaria como son: edad temprana de diagnóstico, multiplicidad o historia familiar de la enfermedad. Por ello, el estudio de estos casos es fundamental para la identificación de nuevos genes que puedan estar relacionados con la patogénesis de la enfermedad.
Mediante técnicas de secuenciación de última generación, el grupo de Cáncer Endocrino Hereditario del Centro Nacional de Investigaciones Oncológicas (CNIO) ha conseguido, a lo largo de estos últimos años, identificar nuevos genes de susceptibilidad a desarrollar PPGL como es el caso de MAX (Comino-Mendez et al, 2011) o MDH2 (Cascon et al 2015). Este último gen, al igual que gran parte de los genes causantes de la enfermedad (SDHA, SDHB, SDHC, SDHD, SDHAF2, IDH1 y FH), pertenece a la ruta central del metabolismo aeróbico celular, el ciclo de Krebs. Los tumores con mutaciones en genes que codifican para enzimas de esta ruta presentan una disrupción de la misma con la consiguiente acumulación de distintos metabolitos. Es sabido que la acumulación de estos intermediarios metabólicos (“oncometabolitos”) tiene consecuencias dramáticas en la epigenética celular, ya que desencadena una serie de modificaciones en la metilación de ciertas regiones del ADN (islas CpG) y de algunas proteínas (histonas) mediante la inactivación de las enzimas encargadas de eliminar los grupos metilo del ADN (TETs) y de las histonas (KDMs) (Letouze et al, 2013). En definitiva, la acumulación de estos “oncometabolitos” origina un fenotipo hipermetilador característico (CIMP, del inglés CpG island methylator phenotype) que provoca a su vez una expresión alterada de un gran número de genes.
El pasado mes de julio, la revista Clinical Cancer Research publicó un estudio (Remacha et al 2017) en el que nuestro grupo demostraba la implicación de dos nuevos genes relacionados con el ciclo de Krebs en el desarrollo de PPGLs.
Los pacientes incluidos en el trabajo se seleccionaron entre aquellos sin mutaciones en los genes conocidos y de acuerdo a la expresión tumoral del gen RBP1, considerado como un marcador de hipermetilación en PPGLs y por lo tanto un indicador de una posible mutación en algún gen del ciclo de Krebs. Los casos finalmente seleccionados se analizaron a través de un panel de secuenciación masiva en el cual se incluyeron todos los genes relacionados con la ruta. Además, se obtuvo el perfil de metilación de todos los tumores secuenciados, para confirmar que se trataba efectivamente de tumores con un perfil CIMP.
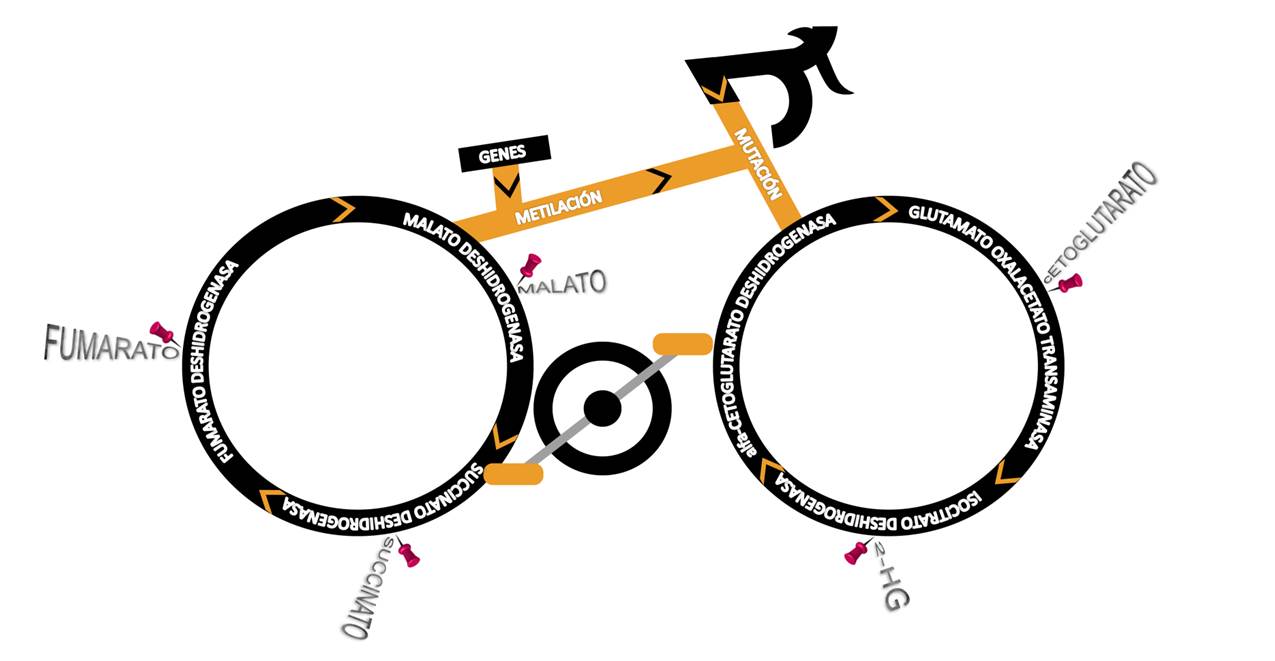
Gracias a esta estrategia, se logró identificar una mutación germinal en el gen GOT2, que codifica una enzima de vital importancia para el intercambio de metabolitos entre la mitocondria y el citosol, en un paciente con múltiples tumores (n=9) y metástasis. El análisis pormenorizado de la variante identificada determinó que era responsable de una mayor expresión del gen en los tumores mutados (tanto de ARNm como de proteína), así como de una mayor actividad enzimática de GOT2 en las células linfoblásticas obtenidas a partir de la sangre del paciente. Además, se observó una acumulación aberrante de succinato en el tejido tumoral mutado y de cetoglutarato tanto en los tumores del paciente como en un modelo celular (basado en células HeLa) en el cual tras silenciar el gen GOT2 se introdujo la proteína con la mutación encontrada en el paciente.
Por otro lado, al aplicar el mismo panel de secuenciación masiva a 60 pacientes con PPGL con el objeto de establecer la frecuencia de mutaciones en el nuevo gen de susceptibilidad identificado, se encontró una mutación germinal truncante en un nuevo gen de susceptibilidad, IDH3B, en un paciente con un único paraganglioma yugular no secretor que presentaba una ratio cetoglutarato/isocitrato alterada.
Por último, en el trabajo también se describen dos mutaciones extremadamente raras en sendos pacientes con PPGL: una metilación somática postzigótica del gen SDHC en un paciente con el síndrome de Carney-Stratakis (múltiples PGLs y GIST), y la tercera mutación conocida hasta la fecha en el gen IDH1 en un paciente con PGL.
Cabe destacar, que la ausencia de mutaciones en algunos de los tumores con perfil CIMP incluidos en el estudio apunta por una parte a la presencia de alteraciones desconocidas en genes relacionados con el ciclo de Krebs, y por la otra a mutaciones en genes implicados en mecanismos de regulación de la metilación del ADN y las histonas (reguladores epigenéticos). Este es el caso del gen H3F3A, en el que se han encontrado recientemente mutaciones somáticas postzigóticas en pacientes con PPGL y tumores óseos (Toledo et al, 2016).
Referencia: Remacha, L., et al., Targeted exome sequencing of Krebs cycle genes reveals candidate cancer predisposing mutations in pheochromocytomas and paragangliomas. Clin Cancer Res, 2017.
Bibliografía:
Cascon, A., et al., Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst, 2015. 107(5).
Comino-Mendez, I., et al., Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet, 2011. 43(7): p. 663-7.
Dahia, P.L., Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer, 2014. 14(2): p. 108-19.
Letouze, E., et al., SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell, 2013. 23(6): p. 739-52.
Toledo, R.A., et al., Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas. Clin Cancer Res, 2016. 22(9): p. 2301-10.

